
Abstract
With the completion of the human genome project, single-nucleotide polymorphisms (SNPs) have become the focus of intense study in biomedical research. Polymerase-mediated primer extension has been employed in a variety of SNP assays. However, these SNP assays using polymerase without proofreading function are compromised by their low reliability. Using a newly developed short amplicon harboring restriction enzyme site, EcoR-I, we were able to compare the single-base discrimination abilities of polymerases with and without proofreading function in primer extension in a broad range of annealing temperatures. Thermodynamic analysis demonstrated a striking single-nucleotide discrimination ability of polymerases with proofreading function. Using unmodified 3′-end allele-specific primers, only template-dependent products were generated by polymerase with proofreading activity. This powerful single-base discrimination ability of exo+ polymerases was further evaluated in primer extension using three types of 3′ terminally modified allele-specific primers. As compared with the poor fidelity in primer extension of polymerases lacking 3′ exonuclease activity, this study provides convincing evidence that the use of proofreading polymerases in combination with 3′-end modified allele-specific primers can be a powerful new strategy for the development of SNP assays.
Similar content being viewed by others


Introduction
Single-nucleotide polymorphisms (SNPs) are the most common form of genetic variation (Sherry et al, 2000; The International SNP Map Working Group, 2001). In the postgenome era, efficient screening of known SNPs is of paramount importance as it can maximize the value of sequence data from the human genome project in critical applications such as fundamental medical research and individualized medicine. At present the high rate of false-positives is one of the major obstacles to the effective application of available high throughput SNP assays, preventing them from being more widely used in clinical applications (Cargill et al, 1999; Chicurel, 2002; Halushka et al, 1999; Lindblad-Toh et al, 2000; Park et al, 2002; Zhang and Li, 2001). A high rate of false-positives occurs in multiple SNP assays mainly as a result of the narrow range of thermal hybridization and wash conditions used to differentiate perfect match from single-base mismatch (Cargill et al, 1999; Chicurel, 2002; Halushka et al, 1999; Lindblad-Toh et al, 2000; Park et al, 2002; Zhang and Li, 2001). Conventional primer extension and oligonucleotide hybridization are restricted in detecting specifically selected SNPs under strictly optimized conditions (Cargill et al, 1999; Halushka, 1999; Higasa and Hayashi, 2002; Lindblad-Toh et al, 2000; Mhlanga and Malmberg, 2001; Park et al, 2002; Tong and Ju, 2002). In the parallel analysis of multiple SNPs, however, the high heterogeneity of different SNPs greatly counteracts the accuracy of currently available assays. To facilitate the practical screening of SNPs, both national and international organizations are urgently requiring the development of new technologies for effectively screening known SNP sites (Chicurel, 2002; The International SNP Map Working Group, 2001).
Although at least more than 20 different methods have been developed for SNP screening, none of them is sufficiently accurate or efficient for clinical application (Beaudet et al, 2001; Chicurel, 2002; DelRio-LaFreniere and McGlennen, 2001; Fei and Smith, 2000; Higasa and Hayashi, 2002; Huang et al, 1992; Kwok et al, 1990; Mhlanga and Malmberg, 2001; Nurmi et al, 2001; Pastinen et al, 2000; Prince et al, 2001; Shi, 2001; Taylor et al, 2001; Tonisson et al, 2002; Zhang and Li, 2001). Among these assays, strategies using polymerase-mediated primer extension are still the major approach used to develop SNP screening methods. These methods include the standard allele-specific PCR followed by single-strand conformational polymorphism, 3′-allele-specific primer extension, single-base extension with ddNTPs, 5′ fluorescently labeled primers using capillary electrophoresis, real-time PCR, and the recently developed primers modified with locked nucleic acid. The large number of SNP assays developed with 3′-mismatched primers reflects the potential of this approach in identification of SNPs. Surprisingly, one neglected or poorly understood area of SNP analysis has been the role played by the 3′-exo proofreading activity. It has been well known for decades that polymerases with proofreading activity have higher fidelity in DNA chain elongation. It is therefore valuable to compare the advantages and disadvantages of polymerases with and without proofreading function in SNP assay.
This study was designed to evaluate the potential use of proofreading polymerases in single-base discrimination. We focus on the behavior of proofreading at the initiation step of primer extension and on how to convert the base-discrimination ability of proofreading into a conveniently available assay for SNP screening. We first studied the thermodynamics of proofreading unmodified 3′ mismatched primers using an amplicon harboring an EcoR-I site. We further tested whether proofreading is able to differentiate isotopically and fluorescently labeled as well as phosphorothioate-modified 3′ terminally mismatched primers.
Results
Leakage Products from Polymerase without 3′ Exonuclease
Figure 1 shows the results from a gradient primer extension using the amplicon with intact EcoR-I site. At an annealing temperature of 63.6° C or lower, 8 out of 12 annealing temperature points had primer-extended products from 3′ terminal mismatched primers similar to those of the perfect-matched primers. The unexpected products were from leakage of the off-switch controlled by 3′ terminal mismatched nucleotide, which was confirmed with subsequent EcoR-I digestion and sequencing analysis. All eight primer-extended products were resistant to EcoR-I digestion and kept the mismatched nucleotide introduced from the primers.

Extension of mismatched and perfectly matched primers by exo− polymerase at various annealing temperatures. (A) Products from perfectly matched primers. (B and C) Primer-extension products with exo− polymerase and mismatched nucleotide at or next to (−2nt) the 3′ terminus of the primer; annealing temperatures between 49 and 63.6° C. At annealing temperatures higher than 63.6° C, exo− polymerase generated product with only the perfect-match primer; no product was generated with the 3′ terminal mismatched primers. Lanes 1, 3, 5, 7, 9, 11, 13, 15, 17, and 19: primer-extension products at annealing temperatures of 49, 49.5, 50.6, 52.2, 54.4, 57.5, 60.9, 63.6, 65.8, and 67.1° C, respectively. Lanes 2, 4, 6, 8, 10, 12, 14, 16, 18, and 20 are the corresponding primer-extension products following EcoR-I digestion. All products from perfectly matched primers were EcoR-I digestible. None of the products from the mismatched primers was digestible by EcoR-I.
With increasing annealing temperatures, exo− polymerase had primer-extension products from perfect-matched primer but failed to extend the 3′ mismatched primer. This base differentiation ability at optimal reaction conditions was in accordance with those of SNP assays using similar mechanisms. However, the unique requirement of optimization of individual SNP sites prevents the exo− polymerase from parallel analysis of multiple SNPs simultaneously. Regarding the target to be analyzed, the leaking products were false-positives because they kept the mismatched nucleotide from the primers and thus failed to identify the base differences of the target (Fig. 1A). In high throughput assay, more or less of the primer-extended products from exo− polymerase will be false-positive from this leakage mechanism as it is impossible to have a universal optimal reaction condition for the large number of different SNP amplicons.
A primer with a mismatch at nucleotide (−2nt) next to its 3′ was used to repeat the primer extension with the same experimental conditions as used above. Similar to the 3′ terminal mismatched primer, exo− polymerase had primer extended products from many of the annealing temperatures tested (Fig. 1C). The products from −2nt mismatched primer were not digestible by EcoR-I either.
To serve as SNP templates, three primer-extension products from 3′ terminal mismatch, −2nt mismatch, and perfect-match primers at annealing temperatures of 49.5 and 58.9° C were also sequenced (Fig. 2). Sequencing data showed that the primer-extended products from perfect-match primer kept the right sequence at the EcoR-I site, GAATTC, and the products from mismatched primer maintained the mismatched sequences of GAATTG and GAATAC for 3′ terminal and −2nt mismatched primers, respectively.

Original sequencing data of primer extension products are shown. The products from perfect-match primer maintain the correct sequence of the EcoR-I site, GAATTC. The products from the mismatched primers inherited the mismatched nucleotide from the respective primers as underlined.
Single-Base Discrimination by Proofreading
Two DNA templates were used differing from each other by one nucleotide: DNA1 containing the wild-type EcoR-I sequence, …GAATTC…, and DNA2 harboring the point-mutated EcoR1 sequence of …GAATTG…. Both templates were analyzed by allele-specific primers using polymerase with proofreading function. As shown in Figure 3, the primer-extension products from the amplicon containing a wild-type sequence of the EcoR-I site was digestible by this enzyme when either the perfect match or the 3′ terminally mismatched allele-specific primer was used. However, the amplicon harboring a single-base mutated sequence of the EcoR-I site was not digestible by EcoR1 regardless of whether the perfect match or the 3′ mismatched allele-specific primer was used. These data clearly show that the 3′ terminal mismatched nucleotide was removed by the proofreading function when the 3′ terminal mismatched primers were used. For 3′ allele-specific primers, polymerases with proofreading function yield template-dependent products, whereas mismatched nucleotides within the primer were retained in the products amplified by polymerases without proofreading activity.

Single-nucleotide identification with 3′ terminal mismatched primer extension by Deep Vent (New England Biolabs, Beverly, MA), a polymerase with proofreading function. Deep Vent efficiently generated products having sequences identical to the template. EcoR-I digestion of the primer-extension products indicates that the 3′ terminal mismatched nucleotide was removed before the primer was extended. Lanes 1 and 5: products from perfect-match primers. Lanes 3 and 7: products from mismatched primers. Lanes 2, 4, 6, and 8: the same products as in 1, 3, 5, 7, respectively, but after EcoR-I digestion.
The 3′ terminal-label primer-extension strategy will be generally of more interest in SNP assay development given that it can be employed without the requirement for enzyme restriction sites. With the use of 3[H]-labeled primer, high isotopic signal was detected (1786 cpm) from products when there was a perfect match between primer and template. This indicates that about 5.7% (1,786/31,000 cpm) of isotopic label was incorporated into the extension products. However, when there was a single-base mismatch between the template and the 3′ terminal of the primer, the signal from extension products was as low as the value for background (91 cpm versus background value of 62 ± 19 cpm, mean ± sd from four blank controls). This significant difference demonstrates that a single-base mismatch can effectively trigger proofreading. This result also suggests the possibility of using a signal-detection method for single-base discrimination without the requirement for enzymatic digestion or gel electrophoresis. Similar to isotopically labeled primer, exo+ polymerase only yields Rox-labeled products when there was a perfect match between the primer and its template. At 1:20 dilution of the original PCR reactions, the matched products had a peak area value of 3685, whereas product yields between mismatched primer and its template showed no Rox-labeled signal when primer extension was done with × 0.4 deoxyribonucleoside triphosphate (dNTP) (80 μm). Using standard dNTP concentrations (200 μm) resulted in very low product yields when there was a mismatch between primer and its template.
With an amplicon containing a single-base mismatch between the 3′ terminus of the 3′ phosphorothioate-modified primer and the template (ie, template harboring the sequence 3′-cttaac-5′), DNA polymerases with and without proofreading function had very different effects on primer extension. As a control Deep Vent (exo−) (New England Biolabs, Beverly, Massachusetts), a DNA polymerase-lacking proofreading function, efficiently yields primer-extended products from 3′ mismatched primers at an annealing temperature of 62.8° C or lower. This was similar to our previous observation using unmodified primers amplified by exo− polymerases (Zhang et al, 2003). When applied to practical SNP assays, the polymerization from mismatched primers might be the major source of false-positives.
A breakthrough phenomenon was observed when using a proofreading polymerase in combination with 3′-terminus-phosphorothioate-modified, 3′-terminus-mismatched primer: the primers with phosphorothioate-modified 3′ termini were not extended at any annealing temperature within the range tested. With the matched amplicon, 3′ terminal phosphorothioate-modified primers were well extended as no enzymatic excision by 3′ exonuclease was required. These data illustrate a perfect on/off switch in DNA polymerization in primer extension with exo+ polymerases having proofreading activity (Table 2). DNA polymerization was turned on when primer and template were perfectly matched and was turned off when there was a single-base mismatch between the phosphorothioate-modified primer's 3′ terminus and the template. Using either short artifical amplicons or natural genomic DNA templates, this data strongly indicates the potential of phosphorothioate-modified primers in practical SNP assays.
Discussion
In this study significant differences were described between the single-base discrimination abilities of polymerases with and without proofreading function. Using a short amplicon harboring an EcoR-I site, proofreading activity was easily evaluated by EcoR-I digestion of the extended products. Over a broad range of annealing temperatures, polymerases with proofreading activity yielded template-dependent products, whereas mismatched nucleotides were retained in the products amplified by polymerases without proofreading activity. The striking single-base discrimination of polymerases with 3′ exonuclease was further tested using 3′-end–labeled primers. The latter makes possible the detection of labeled products without the need for gel electrophoresis.
The proofreading function efficiently removed the labels from mismatched primers. Only products from matched primer led to strong detection signals. When used with exo+ polymerases, 3′ phosphorothioate-modified primers offer very significant additional advantages in the identification of single-base polymorphisms, ie, primer extension turned on when primer and template were perfectly matched and it was turned off when there was a single-base mismatch at the primer's 3′ phosphorothioate-modified terminus. The on/off proofreading phenomenon observed when using exo+ polymerases in combination with 3′ phosphorothioate-modified primers has great potential for SNP analysis.
With polymerases having 3′ to 5′ exonuclease, we demonstrated that 3′ terminal mismatched primers are easily distinguished over a very broad range of reaction conditions as demonstrated by follow-up digestion with restriction endonuclease EcoR-I. The choice of EcoR-I for this study was based entirely on this enzyme's high efficiency. EcoR-I digestion provides an unambiguous (yes or no) readout with which to assess, using our artificial amplicons model, the effectiveness of polymerases with 3′ to 5′ exonuclease activity in single-base discrimination studies. However, although it is possible to apply enzymatic digestion to distinguish the primer-extended products for assaying certain SNP sites, it is obviously impossible to use enzymatic detection when high throughput assays of a wide variety of different SNP sites is required. Another weakness of enzymatic digestion in single-base discrimination is that many SNPs do not create new restriction enzyme sites or the new restriction enzyme sites created are linked to very weak enzymes.
We previously suggested a strategy for the development of novel SNP assays using DNA polymerase with proofreading activity together with labeled 3′ terminal primers (Zhang and Li, 2001). As shown with the artificial amplicon harboring an EcoR-I site, polymerases with proofreading activity in combination with 3′ allele-specific primers can distinguish single-base differences over a broad range of annealing temperatures. From a technical point of view, a more important question is how to generally apply this striking ability of SNP discrimination by proofreading. To address this question, we tested 3′ terminal 3[H]-labeled and fluorescent-labeled primers in single-base discrimination by proofreading. Similar to the EcoR-I digestion strategy, labeled terminal nucleotides provide an unambiguous readout (yes or no) in single-nucleotide discrimination assayed with exo+ polymerases. The 3′ terminal mismatched nucleotide that bears the signal to be detected was removed by the proofreading function, whereas the label was retained in the products when primer and template were perfectly matched. The difficulty in removing Rox-labeled 3′ terminal nucleotide under standard PCR conditions suggests interference by the label on enzymatic digestion by the 3′ exonuclease. The absence of primer extension from mismatched Rox-labeled primer simply indicates a premature termination of DNA polymerization. This was definitively confirmed through the use of 3′ phosphorothioate-modified mismatched primers. Moreover, 3′ phosphorothioate-modified primers in combination with exo+ polymerases demonstrates an attractive on/off switch system for identification of matched and mismatched primers. This is also very promising for the development of practical methods for SNP analysis. Although the underlying mechanism by which the on/off switch operates still remains to be elucidated, the exonuclease-resistant property of the phosphorothioate modification very likely plays a major role as it may delay or block the mismatch removal process mediated by the 3′ exonuclease activity of the polymerase.
Altogether, thermodynamic analysis using EcoR-I digestion demonstrates the compatibility of SNP assay by proofreading with the high throughput screening of heterogeneous templates. An assay that can identify SNPs over a wide temperature range should be readily adaptable to high throughput SNP screening under a single standardized set of reaction conditions. The use of isotopically labeled or fluorescently labeled primers provides a detection method that requires neither enzymatic digestion nor gel electrophoresis. Efficient removal of labeled mismatched nucleotide by the proofreading function of exo+ polymerase renders possible the implementation of assays for sensitive discrimination of single-nucleotide differences. Clearly the use of polymerases with proofreading activity in combination with 3′ allele-specific primers represents a viable and efficient approach for SNP analysis.
A somewhat surprising but reasonable result from this study is that polymerases without proofreading activity do not require stringent Watson-Crick base pairing at the initial step of primer extension, which explains the weakness of these polymerases in SNP assays. On the other hand, this piece of data suggests the possibility of using 3′ mismatched primers for in vitro mutagenesis. In recent years oligonucleotide-mediated mutagenesis has been widely used in gene functional analysis (Hill et al, 1987; Kunkel et al, 1987; Wells et al, 1985). The currently available methods for introducing mutations through the 5′-end of the primer are able to generate several kinds of mutations but are mostly used for point mutations and require follow-up ligation and at least two separate primer-extension reactions (Hill et al, 1987; Kunkel et al, 1987; Wells et al, 1985). The new mutagenesis method using 3′ mismatched primer is simpler and can produce SNP templates in a one-step procedure.
So far, more than 1 million human SNPs have been identified. These SNPs are distributed in the population at very low frequencies (Sherry et al, 2000; The International SNP Map Working Group, 2001). It is easy to amplify some SNPs from any individual, but it is impossible to obtain all of the SNPs from a limited number of individuals because of the low frequencies of many SNPs. To screen for known SNPs, it is very helpful to have the set of SNP templates of interest for use in developing the new assay. The ability to introduce point mutation with 3′ terminal mismatched primers is thus ideal for the preparation of SNP templates. Mutagenesis with 3′ terminal mismatched primers has an additional advantage in genetic analysis in cases where mutation is needed for genes of known sequence. In other words, compared with the 5′ mutation method, this new strategy is less likely to introduce unwanted mutations as a result of errors during primer synthesis. It will be recalled that chemical synthesis of oligonucleotides begins at the 3′-end, and this end is, therefore, less likely to harbor accidental mutations. In other words, the sequences near the 3′ are more reliable than those at the 5′-end as a consequence of their proximity to the start of the synthetic chemical reaction. Reverse phosphoramidites, which allow the chemical synthesis of oligonucleotides beginning at their 5′-ends, might be a remedy to this situation. However, this technology is not yet routinely available (Marin and Zhang, 2000).
In summary this study illustrates different applications of DNA-dependent polymerases with and without proofreading activity for SNP analysis. Polymerases with proofreading activity remove the mismatched nucleotide from the primer before it is extended. The efficient removal of the 3′ terminal nucleotide by the proofreading activity of exo+ polymerases represents a potential new tool for the development of SNP assays. Furthermore, successful removal of 3′ isotopic or fluorescent-labeled mismatched nucleotides during primer extension constitutes a signal-detection system requiring no restriction enzyme digestion or gel electrophoresis and could be immediately applied to SNP high-throughput screening. The observed on/off switch with 3′ phosphorothioate-modified primers and exo+ polymerases further suggests the applicability of these polymerases in the development of SNP assays. This study demonstrates that primer-dependent products are responsible for the high rate of false-positives generated in SNP assays that use allele-specific primers extended by polymerases lacking 3′ exonuclease activity. Although polymerases lacking 3′ exonuclease activity are compromised in single-base discrimination, we demonstrated that their use in 3′ terminal mismatched primer extension is a useful method for introducing mutations into defined sequences. The latter is an especially convenient way to prepare SNP templates.
Materials and Methods
Non-isotopic primers were synthesized commercially by Sengon Inc., (Shanghai, China) and MWG Biotech AG (Charlotte, North Carolina). The 3′ terminal 3[H]-labeled primer was from Genomapping, Inc. (Tianjin, China). Deep Vent and Deep Vent (exo−) were purchased from New England Biolabs, Inc. The amplicons used for single-base discrimination assay were products of Genomapping, Inc.
Experiment 1: Extension of Unmodified Primers by Exo− Polymerase
Deep Vent(exo−) (New England Biolabs, Inc.), a polymerase without 3′ to 5′ exonuclease activity was employed in a two-directional primer extension in annealing temperatures ranging from 46 to 66° C using a short amplicon from mouse renin promoter region with sense primer of TCCCAAGATATCTGAGAATTC and antisense primer of CAGTCTCTAGTTGTGCGGTAAGAAAT. Two additional primers TCCCAAGATATCTGAGAATTG and TCCCAAGATATCTGAGAATAC, mismatched at its 3′ and next to its 3′ terminal nucleotide, were also used. The 12 annealing temperatures used were 46, 46.6, 47.7, 49.5, 52, 55.2, 58.9, 62, 64.5, and 66° C. Following denaturation at 95° C for 2 minutes, primer extension was carried out for 30 cycles as follows: 30 seconds denaturation at 95° C, 30 seconds annealing, and 30 seconds extension at 72° C.
Experiment 2: Extension of Unmodified Primers by Exo+ Polymerase
The EcoR-I restriction site of the short amplicon system was targeted for single-base discrimination with primer extension by polymerase having 3′ to 5′ exonuclease activity (Table 1). The primer extension in a volume of 30 μl was performed with exo+ polymerase at an annealing temperature of 49.5 and 58.9° C for 30 cycles. One-third of the primer-extended product was digested with EcoR-I before gel electrophoresis.
Experiment 3: Extension of 3′ Terminal-Labeled Primers by Exo+ Polymerase
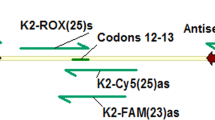
To test the possibility of signal detection without restriction enzyme digestion and gel electrophoresis, primer extension was performed using a 3′ terminal 3[H]-labeled sense primer, TCCCAAGATATCTGAGAATT, and Rox-labeled sense primer, TCCCAAGATATCTGAGAATTC. The corresponding wild-type amplicons and templates with single-base mutations from Experiment 1 were used. The point-mutated templates form a T:T single-base mismatch at the 3′-termini of 3[H]-labeled primer. They also form a C:C single-base mismatch with 3′-end Rox-labeled primer. Primer extension was performed in a total volume of 25 μl, with exo+ polymerase at an annealing temperature of 58.9° C for 30 cycles. A total of 31,000 cpm hot primers was included in the 25-μl final reaction system. Primer-extended products were counted after Qiagen PCR column purification. Primer extension with Rox-labeled primer was performed at an annealing temperature of 53.6° C for 40 seconds with × 1 and × 0.4 dNTP for 30 cycles; results were analyzed using ABI-3100 with Genescan 3.7.
Experiment 4: Extension of Exonuclease-Resistant Primers by Exo+ Polymerase
The 3′ phosphorothioate-modification renders primers resistant to exonuclease digestion. Mismatch removal was well recognized to be the mechanism by which exo+ polymerase maintains the high fidelity of DNA replication. It is interesting to study the effect of 3′ phosphorothioate modification on primer extension between polymerases with and without 3′ exonuclease. As mentioned above we used a short artificial amplicon that harbors an EcoR-I site. In addition, another two single nucleotide targets were analyzed directly using genomic DNA extracted from cultured macrophages (Table 2). Both match and mismatched primers to each template were 3′ phosphorothioate modified. The antisense primers were unmodified primers. The amount of genomic DNA used was 10 ng/μl. Primer extension was carried out at an annealing temperature of 58.9° C for 30 cycles.
References
Beaudet L, Bedard J, Breton B, Mercuri RJ, and Budarf ML (2001). Homogeneous assays for single-nucleotide polymorphism typing using AlphaScreen. Genome Res 11: 600–608.
Cargill M, Altshuler D, Ireland J, Sklar P, Ardlie K, Patil N, Shaw N, Lane CR, Lim EP, Kalyanaraman N, Nemesh J, Ziaugra L, Friedland L, Rolfe A, Warrington J, Lipshutz R, Daley GQ, and Lander ES (1999). Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nat Genet 22: 231–238.
Chicurel M (2002). Faster, better, cheaper genotyping. Nature 412: 580–582.
DelRio-LaFreniere SA and McGlennen RC (2001). Simultaneous allele-specific amplification: A strategy using modified primer-template mismatches for SNP detection–application to prothrombin 20210A (factor II) and factor V Leiden (1691A) gene mutations. Mol Diagn 6: 201–209.
Fei Z and Smith LM (2000). Analysis of single nucleotide polymorphisms by primer extension and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Rapid Commun Mass Spectrom 14: 950–959.
Halushka MK, Fan JB, Bentley K, Hsie L, Shen N, Weder A, Cooper R, Lipshutz R, and Chakravarti A (1999). Patterns of single-nucleotide polymorphisms in candidate genes for blood-pressure homeostasis. Nat Genet 22: 239–247.
Higasa K and Hayashi K (2002). Ordered catenation of sequence-tagged sites and multiplexed SNP genotyping by sequencing. Nucleic Acids Res 30: E11.
Hill DE, Oliphant AR, and Struhl K (1987). Mutagenesis with degenerate oligonucleotides: An efficient method for saturating a defined DNA region with base pair substitutions. Methods Enzymol 155: 558–568.
Huang MM, Arnheim N, and Goodman MF (1992). Extension of base mispairs by TAQ DNA polymerase: Implications for single nucleotide discrimination in PCR. Nucleic Acids Res 20: 4567–4573.
Kunkel TA, Roberts JD, and Zakour RA (1987). Rapid and efficient site-specific mutagenesis without phenotypic selection. Methods Enzymol 154: 367–382.
Kwok S, Kellogg DE, McKinney N, Spasic D, Goda L, Levenson C, and Sninsky JJ (1990). Effects of primer-template mismatches on the polymerase chain reaction: Human immunodeficiency virus type 1 model studies. Nucleic Acids Res 18: 999–1005.
Lindblad-Toh K, Winchester E, Daly MJ, Wang DG, Hirschhorn JN, Laviolette JP, Ardlie K, Reich DE, Robinson E, Sklar P, Shah N, Thomas D, Fan JB, Gingeras T, Warrington J, Patil N, Hudson TJ, and Lander ES (2000). Large-scale discovery and genotyping of single-nucleotide polymorphisms in the mouse. Nat Genet 24: 381–386.
Marin G and Zhang P (2000). DNA Chips/Microarrays and Chemistry. Pharmaceuticals 2000 Web Conference. Available at: http://www.cruachem.co.uk/pages/micro.htm.
Mhlanga MM and Malmberg L (2001). Using molecular beacons to detect single-nucleotide polymorphisms with real-time PCR. Methods 25: 463–471.
Nurmi J, Wikman T, Karp M, and Lovgren T (2001). High-performance real-time quantitative RT-PCR using lanthanide probes and a dual-temperature hybridization assay. Anal Chem 74: 3525–3532.
Park SJ, Taton A, and Mirkin CA (2002). Array-based electrical detection of DNA with nanoparticle probes. Science 295: 1503–1506.
Pastinen T, Raitio M, Lindroos K, Tainola P, Peltonen L, and Syvanen AC (2000). A system for specific, high-throughput genotyping by allele-specific primer extension on microarrays. Genome Res 10: 1031–1042.
Prince JA, Feuk L, Howell WM, Jobs M, Emahazion T, Blennow K, and Brookes AJ (2001). Robust and accurate single nucleotide polymorphism genotyping by dynamic allele-specific hybridization (DASH): Design criteria and assay validation. Genome Res 11: 152–162.
Sherry ST, Ward M, and Sirotkin K (2000). Use of molecular variation in the NCBI dbSNP database. Hum Mutat 15: 68–75.
Shi MM (2001). Enabling large-scale pharmacogenetic studies by high-throughput mutation detection and genotyping technologies. Clin Chem 47: 164–172.
Taylor JD, Briley D, Nguyen Q, Long K, Iannone MA, Li MS, Ye F, Afshari A, Lai E, Wagner M, Chen J, and Weiner MP (2001). Flow cytometric platform for high-throughput single nucleotide polymorphism analysis. Biotechniques 30: 661–669.
The International SNP Map Working Group (2001). A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 409: 928–933.
Tong AK and Ju J (2002). Single nucleotide polymorphism detection by combinatorial fluorescence energy transfer tags and biotinylated dideoxynucleotides. Nucleic Acids Res 30: E19.
Tonisson N, Zernant J, Kurg A, Pavel H, Slavin G, Roomere H, Meiel A, Hainaut P, and Metspalu A (2002). Evaluating the arrayed primer extension resequencing assay of TP53 tumor suppressor gene. Proc Natl Acad Sci USA 99: 5503–5508.
Wells JA, Vasser M, and Powers DB (1985). Cassette mutagenesis: An efficient method for generation of multiple mutations at defined sites. Gene 34: 315–323.
Zhang J and Li K (2001). The 3′ terminal labeled primer extension: A new method of high throughput screening for SNP analysis. Curr Drug Discov 9: 21–24.
Zhang J, Li K, Deng Z, Lao D, Fang W, and Zhang X (2003). Efficient mutagenesis method for producing the templates of single nucleotide polymorphisms. Mol Biotechnol 24: 105–110.
Acknowledgements
We greatly appreciate the free enzyme samples given to us by New England Biolabs, Inc.
This study was partially supported by the National Major Basic Research Program of China (973)(No. G2000056905).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zhang, J., Li, K., Liao, D. et al. Different Applications of Polymerases With and Without Proofreading Activity in Single-Nucleotide Polymorphism Analysis. Lab Invest 83, 1147–1154 (2003). https://doi.org/10.1097/01.LAB.0000081589.91390.DF
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1097/01.LAB.0000081589.91390.DF
