
Abstract
The 26S proteasome constitutes the central proteolytic machinery of the highly conserved ubiquitin/proteasome system, the cell’s major tool for extralysosomal protein degradation. Recently, a plethora of cell proteins implicated in the regulation of basic cellular processes, such as proliferation, differentiation, cell cycling, and apoptosis have been discovered to undergo processing and functional limitation by entering the ubiquitin/proteasome pathway with the final destination to be proteolytically degraded by the 26S proteasome. Because both negative and positive regulators of proliferation and apoptosis undergo proteasomal degradation in a tightly regulated and temporally controlled fashion, the 26S proteasome can play opposite roles in the regulation of proliferation and apoptosis. These roles are apparently defined by the cell’s environment and proliferative state. Finally, proteasomal protein degradation is deregulated in a number of human diseases, including cancer and neurodegenerative and myodegenerative diseases, which all exhibit an imbalance of proliferation and apoptosis. An improved understanding of the modes of proteasomal action should lead to the development of beneficial therapeutic and diagnostic strategies in the future.
Similar content being viewed by others


Main
The discovery of the ubiquitin/proteasome pathway and the characterization of its biologic function as the cell’s major system for extralysosomal protein degradation have pointed out a new conceptual framework for understanding the regulation of basic cellular processes by selective and temporally controlled proteolytic degradation of regulatory proteins (Ciechanover, 1998; Ciechanover et al, 2000; Ciechanover and Schwarz, 1998; Herschko et al, 2000).
In the past few years, a large number of proteins necessary for cell cycle regulation and control of cell proliferation, cell differentiation, and apoptosis (programmed cell death) have been discovered to undergo processing and functional limitation by entering the ubiquitin/proteasome pathway with the final destination to be degraded by the 26S proteasome, a large multicatalytic protease complex (Grimm and Osborne, 2000; Mann and Hilt, 2000; Yew, 2001; Table 1).
To be processed this way, proteins must be targeted for recognition and subsequent degradation by the 26S proteasome by covalent attachments of multiple monomers of the 76 amino acid polypeptide ubiquitin. This process, called ubiquitination, takes place in a multistep reaction governed by three classes of enzymes, namely ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes (E2), and ubiquitin-protein ligases (E3) (Ciechanover et al, 2000).
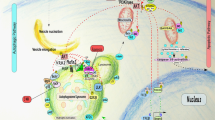
The 26S proteasome, an ATP-dependent multisubunit protease complex found in the cytoplasm and in the nucleus of all eukaryotic cells, constitutes the central proteolytic machinery of the ubiquitin/proteasome system, and is composed of a barrel-shaped 20S catalytic core complex capped at both ends by a 19S regulatory complex (Ferrell et al, 2000; Voges et al, 1999; Fig. 1). The 20S complex has the form of a hollow cylinder composed of four axially stacked rings, each of seven different but related subunits, giving the complex the general stoichiometry of α1–7β1–7β1–7α1–7 (Groll et al, 1997; Löwe et al, 1995). The two inner rings are formed by β subunits, which carry proteolytic sites formed by amino-terminal threonine residues faced to the central cavity of the 20S complex (Baumeister et al, 1998; Voges et al, 1999). The outer rings consist of nonproteolytic α subunits that allow substrate translocation into the central cavity and conformational interactions between the 20S complex and the 19S regulatory complex (Baumeister et al, 1998). The 19S regulatory complex consists of the base subcomplex containing six AAA-ATPases and the lid subcomplex composed of eight non-ATPase subunits that exhibit sequence homologies to human and Arabidopsis COP9-signalosome subunits (Glickman et al, 1998; Henke et al, 1999; Wei et al, 1998). The functions of the 19S regulatory complex seem to be recognition, deubiquitination, unfolding, and translocation of substrate proteins before their proteolytic degradation within the central cavity of the 20S complex (Braun et al, 1999; Glickman et al, 1998; Navon and Goldberg, 2001; Voges et al, 1999; Xie and Varshavsky, 2000).

Subunit composition of and protein degradation by the 26S proteasome (schematic). The 20S catalytic core complex consists of four axially stacked seven-membered rings: two inner rings formed by β subunits (dark blue) carrying proteolytic sites faced to the central cavity of the 20S complex, and two outer rings formed by nonproteolytic α subunits (light brown). The 19S regulatory complex consists of the base subcomplex and the lid subcomplex. The base subcomplex contains six AAA-ATPase subunits (light blue) and two accessory subunits. The lid subcomplex contains eight subunits (yellow). 1, A substrate protein is targeted for proteasomal degradation by the covalent attachment of ubiquitin monomers (red balls). This has been governed previously by ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes (E2), and ubiquitin-protein ligases (E3). 2, The substrate protein enters the 19S regulatory complex, is recognized, deubiquitinated, unfolded, and translocated into the central cavity of the 20S catalytic core complex, where it is degraded by different hydrolytic activities. 3, Short peptides as a product of degradation are released at the opposite end of the 26S proteasome.
Although the occurrence of 26S proteasomes as highly elaborated proteolytic machines is restricted to eukaryotic cells, archaea and bacteria possess proteasomes with less complex subunit compositions (as demonstrated for 20S proteasomes from Thermoplasma acidophilum or Rhodococcus erythropolis and HsIV protease from E. coli), and potentially redundant proteasome-related proteolytic complexes (eg, tricorn protease in T. acidophilum and Streptomyces coelicor) (Baumeister et al, 1998; Tamura et al, 2001; Voges et al, 1999). Thus, protein degradation by the 26S proteasome in eukaryotic cells seems to be a highly conserved process that might have been evolved to fulfill not only metabolic, but also regulatory functions. In fact, basic cellular processes that regulate cell proliferation and apoptosis in eukaryotes are driven by and depend on controlled protein degradation by the 26S proteasome.
Examples of proteasomal involvement in proliferative processes are the regulation of the mitotic cell cycle by temporally controlled proteasomal degradation of cyclins, cyclin-dependent kinases, and their inhibitors (Koepp et al, 1999; Mann and Hilt, 2000; Yew, 2001), and proteasome-dependent regulatory mechanisms in metazoan development that govern oocyte maturation (Josefsberg et al, 2000; Reverte et al, 2001; Sawada et al, 1999; Tokumoto et al, 1999), embryonic cell cycle progression (Josefsberg et al, 2001; Kawahara et al, 2000a, 2000b; Tokumoto et al, 1999), and postembryonic tissue modelling (Dawson et al, 1995; Jones et al, 1995; Löw et al, 1997). Moreover, the 26S proteasome is involved in induction, but also suppression of apoptosis in eukaryotic cells. These opposite functions in the regulation of apoptosis seem to be determined by the proliferative state of a cell and the cellular environment, and may proceed on the basis of the proteasome’s ability to degrade proapoptotic as well as antiapoptotic proteins (Grimm and Osborne, 2000; Orlowski, 1999).
Finally, an increasing body of data provides evidence that deregulation of proteasomal protein degradation contributes to the pathogenesis of human diseases, such as cancer and neurodegenerative and myodegenerative diseases that exhibit an imbalance of proliferation and apoptosis (Chai et al, 1999; Checler et al, 2000; Ciechanover et al, 2000; Cummings et al, 1998; Spataro et al, 1998; Tanaka et al, 2001a). Therefore, the 26S proteasome appears as a regulator of cell proliferation and apoptosis that may govern the delicate balance between a cell’s life and death in dependence on a cell’s biologic state and environment.
Role and Function of the 26S Proteasome in Proliferation
Cell Cycle Control
Proliferation and division of cells implies two basic steps finally yielding to a proper genome duplication: (A) the replication of chromosomal DNA, and (B) the separation and division of sister chromosomes. Both of these steps have to ensure a proper distribution of the entire genome into two new cells:
(A) During the tightly regulated G1/S-phase, the sister chromatids are separated, and complemental DNA synthesis and replication takes place. The proper custodial regulation of DNA replication generally refers to the timely ordered progression from G1- to S-phase that constitutes the strict initiation and completion of only one round of DNA replication in each cell cycle. This duplication relies on the coordinated activities of positive regulators, such as cyclins, cyclin-dependent kinases (CDK), CDK-cyclin complexes, E2F and Cdc6, and negative regulators, such as CDK inhibitors (CKI) of the Cip/Kip and INK4 families. The coordinated timely presence and action of these positive and negative regulators is governed by inactivation as a result of proteasomal degradation (Mann and Hilt, 2000; Yew, 2001).
(B) During the G2/M-phase, the doubled chromosome set is separated along kinetochore microtubules and divided into two new cells. The ordered progression of the S- and M-phase also highly depends on the spatial and temporal control of cell cycle regulatory proteins by proteasomal degradation that finally ensures proper cell cycle transitions and adequate frequencies of cell division (Mann and Hilt, 2000; Tatebe and Yanagida, 2000).
Negative Regulators of Cell Cycle Progression
The following negative regulators of cell cycle progression are known substrates of the 26S proteasome. When these proteins undergo proteasomal degradation, the “brake is released” and cell cycle progression may proceed.
p21Cip1/WAF1.
The CKI p21Cip1/WAF1 is proposed to negatively regulate G1- and early S-phase events through binding to CDKs and/or to proliferating cell nuclear antigen (PCNA) (Luo et al, 1995). In response to DNA damage, the level of p21Cip1/WAF1 protein is dramatically increased as a result of transcriptional activation of p21Cip1/WAF1 by the tumor suppressor protein p53 that finally leads to cell cycle arrest in the G1-phase (El-Deiry et al, 1994). Because selective proteasome inhibitors have been shown to directly induce stabilization of p21Cip1/WAF1 followed by cell cycle arrest (Blagosklonny et al, 1996; Naujokat et al, 2000; Sheaff et al, 2000), p21Cip1/WAF1 is known to be regulated by proteasomal degradation during the cell cycle. Interestingly, proteasomal degradation of p21Cip1/WAF1 in vitro does not strictly require ubiquitination of p21Cip1/WAF1 (Sheaff et al, 2000).
p27Kip1.
Proteasomal degradation of the CKI p27Kip1 is thought to be required for G1-to-S-phase progression (Sutterluty et al, 1999) and mainly occurs at the early onset of S-phase (Tyers and Jorgensen, 2000), although p27Kip1 degradation also can take place at the G0-to-G1-phase transition (Hara et al, 2001). Consequently, p27kip1 protein is abundant in G0 and G1 cells and is down-regulated in proliferating and S-phase cells (Coats et al, 1996). Moreover, ectopic overexpression of mutant p27Kip1, but not of wild-type p27Kip1, results in cell cycle arrest in the S-phase (Sutterluty et al, 1999), strongly suggesting that proteasomal degradation of p27Kip1 is essential for the entry into S-phase. However, inactivation of p27Kip1 function may not only occur by proteasomal degradation, but also via alternative pathways such as proteolytic processing (Shirane et al, 1999).
p19INK4d.
The levels of the CKI p19INK4d have been demonstrated to oscillate periodically during the cell cycle, peaking at S- and G2/M-phase, thereby allowing abundant bindings of p19INK4d to cyclin D-CDK4(6) complexes that result in negative regulation of cell cycle progression (Thullberg et al, 2000). This periodic oscillation of p19INK4d during cell cycle progression is governed by ubiquitination and subsequent proteasomal degradation of p19INK4d, demonstrating that cyclin D-CDK4(6)-dependent cell cycle transitions are regulated by the 26S proteasome (Thullberg et al, 2000).
Geminin.
Geminin is a 25-kd protein that was originally discovered in fertilized Xenopus eggs. Geminin is expressed during the mammalian cell cycle in S-, G2-, and M-phases, but disappears at the time of the metaphase-anaphase transition, where it is ubiquitinated by the anaphase-promoting complex/cyclosome (APC/C), a cell cycle regulated multisubunit ubiquitin-protein ligase, and subsequently degraded by the 26S proteasome (McGarry and Kirschner, 1998). Geminin inhibits DNA replication during S-, G2-, and M-phases by preventing the incorporation of MCM proteins into the prereplication complex. APC/C-triggered proteasomal degradation of geminin at the metaphase-anaphase transition permits DNA replication to proceed in the next cell cycle (McGarry and Kirschner, 1998).
Positive Regulators of Cell Cycle Progression
Positive regulators of cell cycle progression mainly permit the entry into S-phase, and proteasomal degradation of these regulators is thought to tightly control the timely proper entry into S-phase as well as adequate frequencies of S-phase entry.
Cyclin A.
Cyclin A is required for the entry into mitosis and for S-phase progression, and is ubiquitinated and targeted for proteasomal degradation by APC/C during late M- to early G1-phase (Bastians et al, 1999). This suggests that proteasomal degradation of cyclin A is required for the completion of mitosis and the entry into the S-phase of the next cell cycle.
Cyclin D.
Cyclins of the D-type represent a family of growth factor–regulated cyclins that are capable of binding CKIs such as p21Cip1/WAF1 and p27Kip1, resulting in the formation of cyclin-CKI complexes (Cheng et al, 1999). D-type cyclins are required for the entry into S-phase and undergo proteasomal degradation after relocalization to the cytoplasm during S-phase (Diehl et al, 1997). It is thought that the function and abundance of D-type cyclins are not only regulated by proteasomal degradation, but also by cellular localization (Yew, 2001). Defective proteasomal degradation of cyclin D leading to cyclin D accumulation is observed in certain breast cancer cells and is thought to contribute to the neoplastic proliferation of these cells (Russel et al, 1999).
Cyclin E.
Cyclin E is required for the onset of DNA replication during the late G1- and early S-phase, where it is expressed periodically and bound to CDK2 (Ohtsubo et al, 1995). Additionally, unbound forms of cyclin E are present during G1- and S-phase. Both unbound and CDK2-bound cyclin E is targeted for ubiquitination and subsequent proteasomal degradation after its required function at the G1-to-S-phase transition, demonstrating a role for proteasomal degradation in the regulation of S-phase entry (Singer et al, 1999; Winston et al, 1999).
E2F1.
E2F1 is required for the completion of G1-to-S-phase transitions that are thought to be essential for resetting conditions for the next cell cycle (Harper and Elledge, 1999). After its required function at the G1-to-S-phase transition, E2F1 is ubiquitinated and subsequently degraded by the 26S proteasome at S-to-G2-phase (Marti et al, 1999) that is dependent on dissociation of E2F1 from the retinoblastoma tumor suppressor protein Rb (Campanero and Flemington, 1997).
Cdc6.
The Cdc6 protein is a component of the prereplication complex required for initiation of DNA unwinding and replication before S-phase (DePamphilis, 2000). During completion of DNA replication in G1-phase, Cdc6 dissociates from DNA-bound replication complexes and is ubiquitinated and targeted for proteasomal degradation by the APC/C (Peterson et al, 2000).
Development and Differentiation
Development and cellular differentiation in metazoans rely on precise intrinsic programs based on a tightly regulated control of both gene expression and posttranslational protein modification (McGhee, 1995; Morisato and Anderson, 1995; Pahl and Baeuerle, 1996; Zhang et al, 1998). More and more experimental evidence is being collected that the basic molecular events operative in development and differentiation are regulated, at least in part, by the 26S proteasome. For example, early developmental studies in insect model organisms, including Drosophila and Manduca sexta, have suggested that early embryogenesis and metamorphosis in insects depend on proteasome accumulation and tightly regulated proteasomal degradation of certain target proteins (Dawson et al, 1995; Jones et al, 1995; Klein et al, 1990; Löw et al, 1997). Subsequent studies in diverse multicellular organisms, such as sea urchin (Lytechinus pictus), Xenopus laevis, mouse, and rat have demonstrated that developmentally regulated expression of distinct subunits of the proteasomal regulatory 19S subcomplex, as well as proteasomal degradation of cell cycle regulatory proteins, is essential for the initiation of early embryonal mitosis and development (Josefsberg et al, 2001; Kawahara et al, 2000a, 2000b; Tokumoto et al, 1999). Moreover, recent studies in vertebrates reveal that the molecular assembly and the proteolytic activity of certain 26S proteasome subunits undergo changes during the completion of meiosis (oocyte maturation) (Sawada et al, 1999; Tokumoto et al, 1999) and, very recently, it has been demonstrated that the proteolytic activity of the 26S proteasome is absolutely essential for rat and Xenopus oocyte maturation (Josefsberg et al, 2000; Reverte et al, 2001).
Terminal differentiation of eukaryotic cells requires withdrawal from the cell cycle that is accomplished by the down-regulation of CDK activities during G1-phase (Zavitz and Zipursky, 1997). This mainly proceeds, as shown in mammalian cells, through binding of CDKs to the CKIs of the INK4 family (p16Ink4a, p15Ink4b, p18Ink4c, and p19INK4d) and the Cip/Kip family (p21WAF1/Cip1, p27Kip1, and p57Kip2) (Sherr and Roberts, 1999). Because most of these CKIs, including p21WAF1/Cip1, p27Kip1, p57Kip2, and p19INK4d, undergo proteasomal degradation (Blagosklonny et al, 1996; Pagano et al, 1995; Sheaff et al, 2000; Shirane et al, 1999; Thullberg et al, 2000; Urano et al, 1999), the 26S proteasome appears as a negative regulator of terminal differentiation. In fact, this was originally demonstrated in rodent neuronal cells induced to undergo terminal neurite formation by treatment with compounds subsequently identified as specific proteasome inhibitors (Fenteany et al, 1994, 1995; Saito and Kawashima, 1989; Tsubuki et al, 1993, 1996). More recent studies, however, have attempted to define the inhibitory role of the 26S proteasome in differentiation by its degradation of CKIs such as p27Kip1 and p57Kip2. For example, retinoid acid–induced neuronal differentiation of human multipotent embryonal stem cells has been shown to depend on the down-regulation of proteasomal degradation of p27Kip1 (Baldassarre et al, 2000), and transforming growth factor β-induced inhibition of rat osteoblast differentiation has been demonstrated to occur mainly via proteasomal degradation of p57Kip2 (Urano et al, 1999).
By contrast, proteasomal degradation of p21WAF1/Cip1 and other cell cycle regulators, such as cyclin D1 and Cdc25A phosphatase, however, can promote differentiation of certain mouse and human cells (Bernardi et al, 2000; Di Cunto et al, 1998; Negishi et al, 2001; Spinella et al, 1999), ultimately pointing out a complex function of the 26S proteasome in cellular differentiation.
Role and Function of the 26S Proteasome in Apoptosis
Apoptosis is a distinct form of cell death that ensures the control of cell numbers and tissue homeostasis during embryogenesis and postembryonal development and proliferation. The mechanisms of apoptosis are highly conserved from lower eukaryotes to mammals and exhibit a sophisticated network of tightly ordered molecular events that finally converge into the enzymatic fragmentation of chromosomal DNA, thereby driving a cell into death (Hengartner, 2000).
Proapoptotic Functions
First indications of a proapoptotic function of the 26S proteasome came from studies focusing on the developmental regression of abdominal intersegmental muscles of the tobacco hawkmoth, Manduca sexta. Shortly after the emergence of the adult insect from its pupal cuticle, the intersegmental muscles undergo apoptotic regression in response to declining levels of the steroid molting hormone 20-hydroxyecdysone (Schwartz and Truman, 1983). This is accompanied by a dramatic increase in both ubiquitin expression and proteasomal proteolytic activity as evidenced by the 5- to 9-fold enhanced tryptic, chymotryptic, caseinolytic, and peptidylglutamyl-hydrolizing activity of the 26S proteasome (Dawson et al, 1995; Jones et al, 1995). This increase in proteasomal proteolytic activity is correlated with the 8-fold increase in the absolute cellular amounts of the 26S proteasome, the incorporation of four new subunits into the 20S complex, and the 20-hydroxyecdysone–dependent exchange of ATP-ases within the 19S regulatory complex (Dawson et al, 1995; Löw et al, 1997). Such changes in both proteasomal proteolytic activity and subunit composition during development of Manduca sexta show a reprogramming of the 26S proteasome that might result in an enhanced proteasomal degradation of certain ubiquitinated cell proteins, leading to apoptosis of intersegmental muscle cells.
The requirement of proteasomal activity for the progression of apoptosis has been demonstrated originally in two nonproliferating mammalian cell types, resting thymocytes and differentiated neuronal cells (Grimm et al, 1996; Sadoul et al, 1996). Grimm et al were able to demonstrate that primary mouse thymocytes induced to undergo apoptosis by treatment with phorbol 12-myristate 13-acetate, dexamethasone or γ-radiation were rescued from apoptosis when treated with proteasome inhibitors up to 1 hour after the initiation of the apoptotic stimulus. This inhibition of apoptosis was accompanied by prevention of the cleavage of poly(ADP-ribose) polymerase (PARP), which is a biochemical hallmark of caspase activity leading to apoptotic cell death (Hengartner, 2000). By contrast, later addition (3 to 5 hours) of proteasome inhibitors failed to rescue the cells from apoptosis, suggesting that proteasomal activity promotes apoptosis only at upstream points of apoptotic signal transduction pathways. This has been confirmed in later studies showing that proteasome activation occurs upstream of central apoptotic signaling events, such as disruption of mitochondrial transmembrane potential, mitochondrial release of cytochrome c, and activation of caspases in rodent thymocytes induced to undergo apoptosis by various stimuli (Dallaporta et al, 2000; Hirsch et al, 1998; Stefanelli et al, 1998). Similar findings were obtained in studies with differentiated neuronal cells. Sympathetic neurons from rat superior cervical ganglia (Sadoul et al, 1996) and rat cerebellar neurons (Canu et al, 2000) undergoing apoptosis in response to deprivation of nerve growth factor and potassium, respectively, are rescued from apoptosis, and fail to exhibit caspase activation and mitochondrial release of cytochrome c when treated with proteasome inhibitors early after the initiation of the apoptotic stimulus.
One candidate mechanism of how proteasomal activity promotes apoptosis at an upstream point of apoptotic signal transduction has been uncovered recently in primary mouse thymocytes: XIAP and c-IAP1, members of the highly conserved family of inhibitors of apoptosis proteins (IAPs) (Duckett et al, 1996) that exert their antiapoptotic activity, at least in part, by inhibiting the activation and enzymatic activity of caspases (Devereaux et al, 1997, 1998), and by ubiquitination and targeting of caspase-3 for proteasomal degradation (Suzuki et al, 2001), are autoubiquitinated and subsequently degraded by the 26S proteasome in response to various apoptotic stimuli (Yang et al, 2000). This autoubiquitination and subsequent proteasomal degradation of XIAP and c-IAP1 depends on an intact RING finger domain of the proteins, and appears to be highly operative in transducing apoptosis, because cells expressing XIAP and c-IAP1 with mutant RING finger domains display a lack of proteasomal degradation of the mutant proteins and fail to undergo apoptosis induced by diverse stimuli (Yang et al, 2000).
Another candidate mechanism of providing proapoptotic signals by proteasomal activity has been demonstrated in HUVECs induced to undergo apoptosis by treatment with TNF-α. Early after the initiation of TNF-α treatment of HUVECs, Bcl-2, a mitochondrial membrane-anchored protein capable of blocking apoptosis induced by diverse stimuli, was shown to be specifically degraded by the 26S proteasome (Breit-schopf et al, 2000a; Dimmeler et al, 1999). This event was demonstrated to be operative in inducing apoptosis, because pretreatment of HUVECs with specific proteasome inhibitors reversed both TNF-α–induced Bcl-2 degradation and induction of apoptosis (Breit-schopf et al, 2000a; Dimmeler et al, 1999).
Antiapoptotic and Survival Functions
Because early studies revealed that proteasomes are abnormally highly expressed in rapidly growing metazoan embryonic and human neoplastic cells, but not in their well differentiated and normally growing counterparts (Ichihara et al, 1993; Kanayama et al, 1991; Klein et al, 1990; Kumatori et al, 1990; Shimbara et al, 1992), the 26S proteasome has been suggested to play a pivotal role in maintaining survival and proliferation of rapidly and somehow abnormally growing cells (Ichihara and Tanaka, 1995). This hypothesis was supported at the same time by studies in yeast that have shown that disruption of any one of 13 of the 14 genes encoding the subunits of the 20S catalytic core complex leads to complete suppression of cell growth followed by cell death (Fujiwara et al, 1990; Heinemeyer et al, 1991).
The recent identification and availability of selective synthetic and biologic proteasome inhibitors (Fenteany and Schreiber, 1998; Lee and Goldberg, 1998) have allowed a more detailed definition of the roles of the 26S proteasome in fundamental cellular processes, such as apoptosis and proliferation. The stereotyped effects of such proteasome inhibitors observed in neoplastic and rapidly growing cells have finally confirmed the “survival hypothesis” deduced from the early findings of abnormally high proteasome expression in neoplastic and rapidly growing cells: proteasome inhibitors have been found to abundantly induce apoptosis in neoplastic and rapidly growing mammalian cells of hematopoietic (Drexler, 1997; Naujokat et al, 2000; Shinohara et al, 1996; Tanimoto et al, 1997), neuronal (Kitagawa et al, 1999; Lopes et al, 1997; Qiu et al, 2000), mesenchymal (Drexler et al, 2000; Lopes et al, 1997), and epithelial origin (Adams et al, 1999; Herrmann et al, 1998). Intriguingly, human myelogenous leukemic cells abundantly undergoing apoptosis when treated with proteasome inhibitors become refractory to proteasome inhibitor–induced apoptosis after the experimental induction of terminal differentiation leading to noncycling and respectively mature myelogenous cells (Chen et al, 2000; Drexler, 1997). This clearly demonstrates that the proteolytic activity of the 26S proteasome exerts survival functions only in rapidly growing and cycling cells, and also reflects the essential role of the 26S proteasome for maintaining neoplastic proliferation, as already being anticipated in the early studies, which have demonstrated an abnormally high proteasome expression in neoplastic cells, but not in normally growing respective cells (Kumatori et al, 1990; Shimbara et al, 1992).
Numerous cell proteins involved in signal transduction pathways that control proliferation and apoptosis have been discovered to be degraded by the 26S proteasome. Such proteins are supposed to provide a delicate balance of apoptotic and survival signals as a result of their properly regulated levels in normal cells, but might be inappropriately limited or activated in neoplastic cells by enhanced or deregulated proteasomal degradation, thereby allowing neoplastic proliferation.
One such candidate protein is NF-κB, a member of a large family of transcription factors found in the cytoplasm (Verma et al, 1995). NF-κB is sequestered in the cytoplasm by inhibitory IκB proteins and can translocate into the nucleus to act as a transactivator of target genes only after phosphorylation, ubiquitination, and subsequent proteasomal degradation of its inhibitory IκB proteins (Chen et al, 1995; Palombella et al, 1994). NF-κB has been demonstrated to provide antiapoptotic and survival signals in neoplastic cells by transactivation of antiapoptotic genes (Mayo et al, 1997; Wang et al, 1998) and by its high level of activity as a result of deregulated proteasomal degradation of its inhibitory IκB proteins (Besancon et al, 1998; Guzman et al, 2001; Izban et al, 2001; Kordes et al, 2000; Ni et al, 2001). In contrast to this scenario that permits survival signals and suppression of apoptosis as a result of an abundant release of an antiapoptotic protein from its inhibitor, the 26S proteasome is also believed to directly generate antiapoptotic and survival signals in neoplastic cells by degrading proapoptotic proteins such as Bax and Bid (Breitschopf et al, 2000b; Chang et al, 1998; Li and Dou, 2000). Bax and Bid are members of the Bcl-2 family of apoptosis-regulating proteins located in the outer mitochondrial membrane and are involved in the regulation of mitochondrial release of cytochrome c, which is a prerequisite for the execution of apoptosis (Adams and Cory, 1998; Hengartner, 2000).
Another protein found to undergo enhanced and deregulated proteasomal degradation in neoplastic cells is the CKI p27Kip1. p27Kip1, a negative regulator of cell cycle progression involved in differentiation-associated growth arrest (Durand et al, 1997; Wang et al, 1996), is known to be regulated by proteasomal degradation (Pagano et al, 1995; Shirane et al, 1999), which is promoted by binding of p27Kip1 to Jab1, a component of the COP9 signalosome complex homologous to certain subunits of the 19S regulatory proteasomal lid subcomplex (Tomoda et al, 1999). High levels of p27Kip1 as a result of p27Kip1 overexpression or proteasome inhibition can induce apoptosis in tumor cells, ultimately pointing out a proapoptotic and tumor suppressor function of p27Kip1 under certain proliferative conditions (Katayose et al, 1997; Kudo et al, 2000; Schreiber et al, 1999; Wang et al, 1997). Because proteasomal degradation of p27Kip1 is enhanced in several human tumors of epithelial and mesenchymal origin (Chiarle et al, 2000; Esposito et al, 1997; Kawana et al, 1998; Loda et al, 1997; Piva et al, 1999), and low levels of p27Kip1 correlate with tumor progression and poor prognosis (Catzavelos et al, 1997; Chiarle et al, 2000; Sgambato et al, 2000; Slingerland and Pagano, 2000), the loss of p27Kip1 due to deregulated proteasomal degradation may promote the neoplastic proliferation of certain tumor cells.
Other cell cycle regulatory and tumor suppressor proteins, such as p21WAF1/Cip1 and p53, are potential targets for deregulated proteasomal degradation in neoplastic cells. The CKI p21WAF1/Cip1 displays similar properties and functions to those of p27Kip1 (Sherr and Roberts, 1999) and is a known target of proteasomal degradation (Blagosklonny et al, 1996; Sheaff et al, 2000). As in the case of p27Kip1, overexpression of p21WAF1/Cip1 causes induction of apoptosis in tumor cells (Katayose et al, 1995; Schreiber et al, 1999; Sheikh et al, 1995), and accumulation of p21WAF1/Cip1 induced by proteasome inhibition has been shown to precede G2/M arrest and apoptosis in rapidly growing cells (Naujokat et al, 2000; Rieber and Rieber, 2000), suggesting, however, a role for a deregulated proteasomal degradation of p21WAF1/Cip1 in maintaining neoplastic proliferation.
The tumor suppressor protein p53 is a short-lived nuclear protein capable of inducing both growth arrest and apoptosis after its stabilization in response to genotoxic stress and DNA damage (Levine, 1997; Sionov and Haupt, 1999). This stabilization of p53 leads to growth arrest or apoptosis (with the final outcome of tumor suppression) and is thought to occur mainly via the down-regulation of its proteasomal degradation, because inhibition of the ubiquitin/proteasome pathway by various means has been demonstrated to result in p53 stabilization followed by apoptosis (Chowdary et al, 1994; Lopes et al, 1997; Maki et al, 1996; Monney et al, 1998). Apoptosis in such case could well be mediated by a combination of up-regulation of both the genes of Bax and p21WAF1/Cip1, proapoptotic targets of p53 (El-Deiry et al, 1994; Miyashita and Reed, 1995), and by down-regulation of the antiapoptotic protein Bcl-2, which is repressed by p53 (Agarwal et al, 1998). An example of enhanced proteasomal degradation of p53 leading to neoplastic proliferation is well documented in human papilloma virus (HPV)–related cervical carcinomas: the E6 oncoprotein encoded by the high risk HPV-16 and -18 subtypes binds to p53 and promotes its rapid degradation by the 26S proteasome, a feature that is essential for the neoplastic growth of HPV-positive carcinoma cells (Hengstermann et al, 2001; Scheffner et al, 1990). Moreover, it has been demonstrated recently that human chronic lymphocytic leukemia cells exhibit a constitutively up-regulated proteasomal proteolytic activity leading to an enhanced degradation of p53, which is thought to allow survival and neoplastic proliferation of these cells (Masdehors et al, 2000).
Finally, there is increasing evidence for a functional cooperation of the COP9 signalosome, a multimeric protein complex homologous to the 19S regulatory proteasomal lid subcomplex (Henke et al, 1999), with the 26S proteasome in degradation of p53, p27Kip1, IκBα, and c-Jun (Bech-Otschir et al, 2001; Seeger et al, 1998; Tomoda et al, 1999). This cooperation may also contribute to deregulated proteasomal protein degradation in neoplastic cells.
Role of the 26S Proteasome in the Pathogenesis of Human Diseases
An increasing body of recent data provides evidence that deregulation of the ubiquitin/proteasome system contributes to the pathogenesis of several human diseases, such as cancer and neurodegenerative and myodegenerative diseases that exhibit an imbalance of proliferation and apoptosis.
Cancer
In various entities of human cancer, a deregulated proteasomal proteolysis of tumor suppressors and oncoproteins has been associated with initiation and maintenance of neoplastic proliferation as well as tumor progression and poor prognosis. This has been widely demonstrated in the case of enhanced proteasomal degradation of p27Kip1 in lung carcinomas (Catzavelos et al, 1999; Esposito et al, 1997; Kawana et al, 1998), colorectal carcinomas (Loda et al, 1997), breast carcinomas (Catzavelos et al, 1997; Porter et al, 1997), gliomas (Piva et al, 1999), and lymphomas (Chiarle et al, 2000). Moreover, enhanced proteasomal degradation of the tumor suppressor protein p53 is clearly linked to the development and neoplastic proliferation of HPV-related cervical carcinomas (Hengstermann et al, 2001; Scheffner et al, 1990), and is thought to confer resistance to apoptosis in chronic lymphocytic leukemia (Masdehors et al, 2000).
A more indirect function of the 26S proteasome in the permission of cancer might be operative in the multistep carcinogenesis of inherited colorectal carcinomas: the oncoprotein β-catenin, a central component of signaling and differentiation pathways of the colorectal epithelium, is targeted for proteasomal degradation by the tumor suppressor APC (adenomatous polyposis coli), which is mutated or lost in inherited colorectal carcinomas. The consequent accumulation of β-catenin in APC-defective colorectal carcinomas leads to the activation of signaling pathways that promote carcinogenesis and neoplastic proliferation (Aberle et al, 1997; Inomata et al, 1996; Korink et al, 1997; Morin et al, 1997; Powell et al, 1992; Sellin et al, 2001).
Parkinson’s Disease
Parkinson’s disease (PD) is a neurodegenerative movement disorder characterized by the selective cell death of dopaminergic neurons located in the substantia nigra of the midbrain. By its clinical definition, PD represents a heterogenous disorder that encompasses a small proportion of individuals with an autosomal recessive juvenile form of PD (AR-JP) and a larger population with an idiopathic form of PD with the neuropathologic stigma of Lewy bodies, cytoplasmic inclusion bodies consisting of aggregates of abnormally accumulated proteins that are not observed in AR-JP (Hattori et al, 2000; Kitada et al, 2000). α-synuclein, a protein that forms the major fibrillary component of Lewy bodies, has been shown to interact with the 19S proteasomal regulatory complex and to undergo proteasomal degradation (Bennett et al, 1999; Ghee et al, 2000). Intriguingly, proteasomal proteolytic activity is dramatically impaired in dopaminergic neurons in idiopathic PD (McNaught and Jenner, 2001), and recent experimental data provide evidence that proteasome inhibition in α-synuclein–expressing neuronal cells results in accumulation and aggregation of α-synuclein, leading to apoptosis (Rideout et al, 2001; Tanaka et al, 2001b; Tofaris et al, 2001).
The parkin gene, which is implicated in the onset and progression of AR-JP (Kitada et al, 1998), encodes the 53-kd parkin protein that functions as a ubiquitin-protein ligase for targeting proteins for proteasomal degradation (Shimura et al, 2000). Parkin mutations that are frequently found in AR-JP patients (Kitada et al, 2000) lead to the functional loss of the parkin protein as a ubiquitin-protein ligase. This failure causes accumulation of several proteins destined to be degraded by the 26S proteasome, and finally results in apoptosis (Imai et al, 2001; Tanaka et al, 2001a).
Alzheimer’s Disease
Alzheimer’s disease (AD), the most common form of senile dementia, is characterized by the progressive degeneration and loss of cortical and limbic neurons that is associated with the extracellular deposition of amyloid β-peptides in senile plaques and the occurrence of intracellular neurofibrillary tangles composed of paired helical filaments of hyperphosphorylated tau proteins. Because the presence of ubiquitin and distinct proteasomal subunits has been detected immunohistochemically in both senile plaques and neurofibrillary tangles (Fergusson et al, 1996; Ii et al, 1997), it has been postulated that defective proteolytic functions of the 26S proteasome contribute to the pathogenesis of AD. However, subtle biochemical studies have revealed that amyloid β selectively inhibits the proteasomal chymotrypsin-like proteolytic activity in a noncompetitive fashion (Gregori et al, 1995, 1997) and, in turn, proteasomal inhibition leads to an increased α-secretase–dependent production of amyloid β and its precursors (Marambaud et al, 1997, 1998). This multimodal amplification of proteasome inhibition in the context of a pathologic amyloid β metabolism is likely to explain the global impairment of proteasome activity that has been detected postmortally in cortical neurons of AD patients (Keller et al, 2000; Salon et al, 2000). Moreover, a mutant form of ubiquitin, termed Ub(+1), is selectively expressed in AD neurons and competes for ubiquitination of proteasomal target proteins with normal ubiquitin, thereby inhibiting proteasomal protein degradation (Lam et al, 2000). Thus, defective proteasome function may cause, at least in part, the accumulation of aberrant protein aggregates leading to neuronal degeneration and cell death in AD.
Polyglutamine Diseases and Muscle Dystrophy
Polyglutamine diseases encompass an increasing number of inherited neurodegenerative diseases characterized by the expansion of CAG trinucleotide repeats in the causative genes that leads to the translation of polyglutamine stretches abundantly found in the encoded proteins. These abnormal proteins are thought to be involved in the induction of apoptotic mechanisms causing neuronal degeneration in several polyglutamine diseases, such as spinocerebellar ataxias (SCAs) and Huntington’s disease (Evert et al, 2000). Immunohistochemical studies in neuronal tissues from patients with SCA type 1 and SCA type 3 have demonstrated that the 26S proteasome colocalizes with intranuclear inclusions formed by aggregates of the abnormal polyglutamine proteins ataxin-1 and ataxin-3, respectively (Chai et al, 1999; Cummings et al, 1998). These findings have been confirmed recently in a more detailed study showing that an ATPase subunit of the 19S proteasomal regulatory complex specifically interacts with ataxin-7, the abnormal polyglutamine protein found in SCA type 7 (Matilla et al, 2001). Moreover, in ataxin-3–transfected neurons, the 26S proteasome has been shown to redistribute into nuclear inclusions formed by ataxin-3 (Chai et al, 1999). Interestingly, proteasome inhibitors have been found to increase nuclear aggregate formation of polyglutamine proteins in SCA type 3 and Huntington’s disease neurons (Chai et al, 1999; Martin-Aparicio et al, 2001), suggesting a rather protective role of proteasomal activity in polyglutamine diseases.
Catabolic muscle wasting during pathologic conditions associated with cancer cachexia, muscular dystrophies, amyotrophic lateral sclerosis, and peripheral neuropathies has been shown to occur as a result of muscle fiber degradation mainly by the 26S proteasome (Hasselgren et al, 2002; Kumamoto et al, 2000; Williams et al, 1999). However, several accessory mechanisms, including calpain-dependent proteolysis and deregulated activities of tripeptidyl peptidase II and COP9 signalosome, might be involved in the process of proteolytic muscle wasting (Hasselgren et al, 2002).
Conclusions
Proteolytic degradation of cell proteins by the 26S proteasome is a highly complex and tightly regulated process that plays pivotal roles in the regulation of basic cellular processes, including differentiation, proliferation, cell cycling, apoptosis, gene expression, and signal transduction. The mechanisms that underlie the regulation of both proteasomal protein degradation and polyubiquitination of target proteins as a prerequisite for degradation are still largely unknown, and the considerable number of proteasomal target proteins discovered to date may represent only a minor part of the myriad proteins awaiting the identification as proteasomal substrates.
From a mechanistic view, the 26S proteasome is capable of governing strictly opposite biologic features that crucially determine the fate of a cell, proliferation, and apoptosis. Moreover, also within the regulation of proliferation and apoptosis, the 26S proteasome can play opposite roles as revealed by its ability to contribute to either inhibition or promotion of each of these basic cellular processes, apparently in dependence on a cell’s environment and proliferative state. Because proteasomal protein degradation is a highly ordered and elaborated process, it is obvious that this process also can underlie deregulation as observed in several human diseases that exhibit an imbalance of proliferation and apoptosis as a fundamental pathogenetic feature. Future research will lead to an improved understanding of the modes of proteasomal action with the aim of allowing the development of beneficial therapeutic strategies.
References
Aberle H, Bauer A, Stappert J, Kispert A, and Kemler R (1997). β-catenin is a target for the ubiquitin-proteasome pathway. EMBO J 16: 3797–3804.
Adams J, Palombella VJ, Sausville EA, Johnson J, Destree A, Lazarus DD, Maas J, Pien CS, Prakash S, and Elliott PJ (1999). Proteasome inhibitors: A novel class of potent and effective antitumor agents. Cancer Res 59: 2615–2622.
Adams JM and Cory S (1998). The Bcl-2 protein family: Arbiters of cell survival. Science 281: 1322–1325.
Agarwal ML, Taylor WR, Chernov MV, Chernova OB, and Stark GR (1998). The p53 network. J Biol Chem 273: 1–4.
Baldassarre G, Boccia A, Bruni P, Sandomenico C, Barone MV, Pepe S, Angrisano T, Belleti B, Motti ML, Fusco A, and Viglietto G (2000). Retinoic acid induces neuronal differentiation of embryonal carcinoma cells by reducing proteasome-dependent proteolysis of the cyclin-dependent inhibitor p27. Cell Growth Differ 11: 517–526.
Bastians H, Topper LM, Gorbsky GL, and Ruderman JV (1999). Cell cycle-regulated proteolysis of mitotic target proteins. Mol Cell Biol 10: 3927–3941.
Baumeister W, Walz J, Zühl F, and Seemüller E (1998). The proteasome: Paradigm of a self-compartmentalizing protease. Cell 92: 367–380.
Bech-Otschir D, Kraft R, Huang X, Henklein P, Kapelari B, Pollmann C, and Dubiel W (2001). COP9 signalosome-specific phosphorylation targets p53 to degradation by the ubiquitin system. EMBO J 20: 1630–1639.
Bennett MC, Bishop JF, Leng Y, Chock PB, Chase TN, and Mouradian MM (1999). Degradation of α-synuclein by proteasome. J Biol Chem 274: 33855–33858.
Bernardi R, Liebermann DA, and Hoffmann B (2000). Cdc25A stability is controlled by the ubiquitin-proteasome pathway during cell cycle progresssion and terminal differentiation. Oncogene 19: 2447–2454.
Besancon F, Atfi A, Gespach C, Cayre YE, and Bourgeade MF (1998). Evidence for a role of NF-κB in the survival of hematopoietic cells mediated by interleukin 3 and the oncogenic TEL/platelet-derived growth factor receptor β fusion protein. Proc Natl Acad Sci USA 95: 8081–8086.
Blagosklonny MV, Wu GS, Omura S, and El-Deiry WS (1996). Proteasome-dependent regulation of p21WAF1/CIP1 expression. Biochem Biophys Res Commun 227: 564–569.
Braun BC, Glickman M, Kraft R, Dahlmann B, Kloetzel PM, Finley D, and Schmidt M (1999). The base of the proteasome regulatory particle exhibits chaperone-like activity. Nat Cell Biol 1: 221–226.
Breitschopf K, Haendeler J, Malchow P, Zeiher AM, and Dimmeler S (2000a). Posttranslational modification of Bcl-2 facilitates its proteasome-dependent degradation: Molecular characterization of the involved signaling pathways. Mol Cell Biol 20: 1886–1896.
Breitschopf K, Zeiher AM, and Dimmeler S (2000b). Ubiquitin-mediated degradation of the proapoptotic form of Bid. J Biol Chem 275: 21648–21652.
Campanero MR and Flemington EK (1997). Regulation of E2F through ubuquitin-proteasome-dependent degradation: Stabilization by the pRB tumor suppressor protein. Proc Natl Acad Sci USA 94: 2221–2226.
Canu N, Barbato C, Ciotti MT, Serafino A, Dus L, and Calissano P (2000). Proteasome involvement and accumulation of ubiquitinated proteins in cerebellar granule neurons undergoing apoptosis. J Neurosci 15: 589–599.
Catzavelos C, Bhattacharya N, Ung YC, Wilson JA, Roncari L, Sandhu C, Shaw P, Yeger H, Morava-Protzner I, Kapusta L, Franssen E, Pritchard KI, and Slingerland JM (1997). Decreased levels of the cell cycle inhibitor p27Kip1 protein: Prognostic implications in primary breast cancer. Nat Med 3: 227–230.
Catzavelos C, Tsao MS, DeBoer G, Bhattacharya N, Shepherd F, and Slingerland JM (1999). Reduced expression of the cell cycle inhibitor p27Kip1 in non-small cell lung carcinoma: A potential prognostic factor independent of ras. Cancer Res 59: 684–688.
Chai YH, Koppenhafer SL, Shoesmith SJ, Perez MK, and Paulson HL (1999). Evidence for proteasome involvement in polyglutamine disease: Localization to nuclear inclusions in SCA3/MJD and suppression of polyglutamine aggregation in vitro. Hum Mol Genet 8: 673–682.
Chang YC, Lee YS, Tejima T, Tanaka K, Omura S, Heintz NH, Mitsui Y, and Magae J (1998). Mdm2 and bax, downstream mediators of the p53 response, are degraded by the ubiquitin-proteasome pathway. Cell Growth Diff 9: 79–84.
Checler F, da Costa CA, Ancolio K, Chevallier N, Lopez-Perez E, and Marambaud P (2000). Role of proteasome in Alzheimer’s disease. Biochim Biophys Acta 1502: 133–138.
Chen C, Lin H, Karanes C, Pettit GR, and Chen BD (2000). Human THP-1 monocytic leukemic cells induced to undergo monocytic differentiation by bryostatin 1 are refractory to proteasome inhibitor-induced apoptosis. Cancer Res 60: 4377–4385.
Chen ZJ, Hagler J, Palombella V, Melandri F, Scherer D, Ballard D, and Maniatis T (1995). Signal-induced site-specific phosphorylation targets IκBα to the ubiquitin-proteasome pathway. Genes Dev 9: 1586–1597.
Cheng J, Olivier P, Diehl JA, Fero M, Roussel MF, Roberts JM, and Sherr CJ (1999). The p21Cip1 and p27Kip1 CDK inhibitors are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO J 18: 1571–1583.
Chiarle R, Budel LM, Skolnik J, Frizzera G, Chilosi M, Corato A, Pizzolo G, Magidson J, Montagnoli A, Pagano M, Maes B, De Wolf-Peeters C, and Inghirami G (2000). Increased proteasome degradation of cyclin-dependent kinase inhibitor p27 is associated with a decreased overall survival in mantle cell lymphoma. Blood 95: 619–626.
Chowdary DR, Dermondy JJ, Jha KK, and Ozer HL (1994). Accumulation of p53 in a mutant cell line defective in the ubiquitin pathway. Mol Cell Biol 14: 1997–2003.
Ciechanover A (1998). The ubiquitin-proteasome pathway: On protein death and cell life. EMBO J 17: 7151–7160.
Ciechanover A and Schwarz AL (1998). The ubiquitin-proteasome pathway: The complexity and myriad function of proteins death. Proc Natl Acad Sci USA 95: 2727–2730.
Ciechanover A, Orian A, and Schwartz AL (2000). Ubiquitin-mediated proteolysis: Biological regulation via destruction. BioEssays 22: 442–451.
Coats S, Flanagan WM, Nourse J, and Roberts JM (1996). Requirement of p27Kip1 for restriction point control of the fibroblast cell cycle. Science 272: 877–880.
Cummings CJ, Mancini MA, Antallfy B, DeFranco DB, Orr HT, and Zoghbi HY (1998). Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat Genet 19: 148–154.
Dallaporta B, de Pablo M, Maisse C, Daugas E, Loeffler M, Zamzami N, and Kroemer G (2000). Proteasome activation as a critical event of thymocyte apoptosis. Cell Death Diff 7: 368–373.
Dawson SP, Arnold JE, Mayer NJ, Reynolds SE, Billett MA, Gordon C, Colleaux L, Kloetzel PM, Tanaka K, and Mayer RJ (1995). Developmental changes of the 26S proteasome in abdominal intersegmental muscles of Manduca sexta during programmed cell death. J Biol Chem 270: 1850–1858.
DePamphilis ML (2000). Nuclear structure and DNA replication. J Struct Biol 129: 186–197.
Devereaux QL, Roy N, Srennicke HR, van Arsdale T, Zhou Q, Srinivasula SM, Alnemri ES, Salvesen GS, and Reed JC (1998). IAPs block apoptotic events induced by caspase-8 and cytochrome c by direct inhibition of distinct caspases. EMBO J 17: 2215–2223.
Devereaux QL, Takahashi R, Salvesen GS, and Reed JC (1997). X-linked IAP is a direct inhibitor of cell-death proteases. Nature 388: 300–304.
Di Cunto F, Topley G, Calautti E, Hsiao J, Ong L, Seth PK, and Dotto GP (1998). Inhibitory function of p21Cip1/WAF1 in differentiation of primary mouse keratinocytes independent of cell cycle control. Science 280: 1069–1072.
Diehl JA, Zindy F, and Sherr CJ (1997). Inhibition of cyclin D1 phosphorylation on threonine-286 prevents its rapid degradation via the ubiquitin-proteasome pathway. Genes Dev 11: 957–972.
Dimmeler S, Breitschopf K, Haendeler J, and Zeiher AM (1999). Dephosphorylation targets Bcl-2 for ubiquitin-dependent degradation: A link between the apoptosome and the proteasome pathway. J Exp Med 189: 1815–1822.
Drexler HCA (1997). Activation of the cell death program by inhibition of proteasome function. Proc Natl Acad Sci USA 94: 855–860.
Drexler HCA, Risau W, and Konerding MA (2000). Inhibition of proteasome function induces cell death in proliferating endothelial cells. FASEB J 14: 65–77.
Duckett CS, Nava VE, Gedrich RW, Clem RJ, Van Dongen JL, Gilfillan MC, Shiels H, Hardwick JM, and Thompson CB (1996). A conserved family of cellular genes related to the baculovirus iap gene and encoding apoptosis inhibitors. EMBO J 15: 2685–2694.
Durand B, Gao FB, and Raff M (1997). Accumulation of the cyclin-dependent kinase inhibitor p27/Kip1 and the timing of oligodendrocyte differentiation. EMBO J 16: 306–317.
El-Deiry WS, Harper JW, O’Connor PM, Velculescu VE, Canman CE, Jackman J, Pietenpol JA, Burrell M, Hill DE, Wang Y, Wiman KG, Mercer WE, Kastan MB, Kohn KW, Elledge SJ, Kinzler KW, and Vogelstein B (1994). WAF1/CIP1 is induced in p53-mediated G1 arrest and apoptosis. Cancer Res 54: 1169–1174.
Esposito V, Baldi A, De Luca A, Groger AM, Loda M, Giordano GG, Caputi M, Baldi F, Pagano M, and Giordano A (1997). Prognostic role of the cyclin-dependent kinase inhibitor p27 in non-small cell lung cancer. Cancer Res 57: 3381–3385.
Evert BO, Wullner U, and Klockgether T (2000). Cell death in polyglutamine diseases. Cell Tissue Res 301: 189–204.
Fenteany G and Schreiber SL (1998). Lactacystin, proteasome function, and cell fate. J Biol Chem 273: 8545–8548.
Fenteany G, Standaert RF, Lane WS, Choi S, Corey EJ, and Schreiber SL (1995). Inhibition of proteasome activities and subunit-specific amino-terminal threonine modification by lactacystin. Science 268: 726–731.
Fenteany G, Standaert RF, Reichard GA, Corey EJ, and Schreiber SL (1994). A β-lactone related to lactacystin induces neurite outgrowth in a neuroblastoma cell line and inhibits cell cycle progression in an osteosarcoma cell line. Proc Natl Acad Sci USA 91: 3358–3362.
Fergusson J, Landon M, Lowe J, Dawson SP, Layfield R, Hanger DP, and Mayer RJ (1996). Pathological leasons of Alzheimer’s disease and dementia with Lewy bodies brains exhibit immunoreactivity to an ATPase that is a regulatory subunit of the 26S proteasome. Neurosci Lett 219: 167–170.
Ferrell K, Wilkinson CRM, Dubiel W, and Gordeon C (2000). Regulatory subunit interactions of the 26S proteasome, a complex problem. Trends Biochem Sci 25: 83–88.
Fujiwara T, Tanaka K, Ornino E, Yoshimura T, Kumatori A, Tamura T, Chung CH, Nakai T, Yamaguchi K, Shin S, Kakizuka A, Nakanishi S, and Ichihara A (1990). Proteasomes are essential for yeast proliferation. J Biol Chem 265: 16604–16613.
Ghee M, Fournier A, and Mallet J (2000). Rat α-synuclein interacts with Tat binding protein 1, a component of the 26S proteasomal complex. J Neurochem 75: 2221–2224.
Glickman MH, Rubin DM, Coux O, Wefes I, Pfeifer G, Cjeka Z, Baumeister W, Fried VA, and Finley D (1998). A subcomplex of the proteasome regulatory particle required for ubiquitin-conjugate degradation and related to the COP9-signalosome and elF3. Cell 94: 615–623.
Gregori L, Fuchs C, Figueiredo-Pereira ME, Van Nostrand WE, and Goldgaber D (1995). Amyloid β-protein inhibits ubiquitin-dependent protein degradation in vitro. J Biol Chem 270: 19702–19708.
Gregori L, Hainfeld JF, Simon MN, and Goldgaber D (1997). Binding of amyloid β to the 20S proteasome. J Biol Chem 272: 58–62.
Gregory MA and Hann SR (2000). c-Myc proteolysis by the ubiquitin-proteasome pathay: Stabilization of c-Myc in Burkitt’s lymphoma cells. Mol Cell Biol 20: 2423–2435.
Grimm LM and Osborne BA (2000). The role of the proteasome in apoptotsis. In: Hilt W, and Wolf DH, editors. Proteasomes: The world of regulatory proteolysis. Georgetown, Texas: Landes Bioscience, 315–325.
Grimm LM, Goldberg AL, Poirier GG, Schwartz LM, and Osborne BA (1996). Proteasomes play an essential role in thymocyte apoptosis. EMBO J 15: 3835–3844.
Groll M, Ditzel L, Löwe J, Stock D, Bochtler M, Bartunik H, and Huber R (1997). Structure of 20S proteasome from yeast at 2.4 Å resolution. Nature 386: 463–471.
Guzman ML, Neering SJ, Upchurch D, Grimes B, Howard DS, Rizzieri DA, Luger SM, and Jordan CT (2001). Nuclear factor-kappa B is constitutively activated in primitive human acute myelogenous leukemia cells. Blood 98: 2301–2307.
Hara T, Kamura T, Nakayama K, Oshikawa K, Hatakeyama S, and Nakayama KI (2001). Degradation of p27Kip1 at the G0-G1 transition mediated by a Skp2-independent ubiquitination pathway. J Biol Chem 276: 48937–48943.
Harper JW and Elledge SJ (1999). Skipping into the E2F1-destruction pathway. Nat Cell Biol 1: E5–E7.
Hasselgren PO, Wray C, and Mammen J (2002). Molecular regulation of muscle cachexia: It may be more than the proteasome. Biochem Biophys Res Commun 290: 1–10.
Hattori N, Shimura H, Kubo S, Kitada T, Wang M, Asakawa S, Minashima S, Shimizu N, Suzuki T, Tanaka K, and Mizuno Y (2000). Autosomal recessive juvenile parkinsonism: A key to understanding nigral degeneration in sporadic Parkinson’s disease. Neuropathol 20 (Suppl): 85–90.
Heinemeyer W, Kleinschmid JA, Saidowski J, Escher C, and Wolf DH (1991). Proteinase yscE, the yeast proteasome/multicatalytic multifunctional proteinase: Mutants unravel its function in stress induced proteolysis and uncover its necessity for cell survival. EMBO J 10: 555–562.
Hengartner MO (2000). The biochemistry of apoptosis. Nature 407: 770–776.
Hengstermann A, Linares LK, Ciechanover A, Whitaker NJ, and Scheffner M (2001). Complete switch from Mdm2 to human papillomavirus E6-mediated degradation of p53 in cervical cancer cells. Proc Natl Acad Sci USA 98: 1218–1224.
Henke W, Ferrell K, Bech-Otschir D, Seeger M, Schade R, Jungblut P, Naumann M, and Dubiel W (1999). Comparison of human COP9 signalosome and 26S proteasome lid. Mol Biol Rep 26: 29–34.
Herrmann JL, Briones F Jr, Brisbay S, Logothetis CL, and McDonnell TJ (1998). Prostate carcinoma cell death resulting from inhibition of proteasome activity is independent of functional Bcl-2 and p53. Oncogene 17: 2889–2899.
Herschko A, Ciechanover A, and Varshavsky A (2000). The ubiquitin system. Nat Med 6: 1073–1081.
Hirsch T, Dallaporta B, Zamzami N, Susin SA, Ravagnan L, Marzo I, Brenner C, and Kroemer G (1998). Proteasome activation occurs at an early, premitochondrial step of thymocyte apoptosis. J Immunol 161: 35–40.
Ichihara A and Tanaka K (1995). Roles of proteasomes in cell growth. Mol Biol Rep 21: 49–52.
Ichihara A, Tanaka K, Andoh T, and Shimbara N (1993). Regulation of proteasome expression in developing and transformed cells. Adv Enzyme Regul 33: 173–180.
Ii K, Ito H, Tanaka K, and Hirano A (1997). Immunocytochemical co-localization of the proteasome in ubiquitinated structures in neurodegenerative diseases and the elderly. J Neuropathol Exp Neurol 56: 125–131.
Imai Y, Soda M, Inoue H, Hattori N, Mizuno Y, and Takahashi R (2001). An unfolded putative polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of parkin. Cell 105: 891–902.
Inomata M, Ochiai A, Akimoto S, Kitano S, and Hirohashi S (1996). Alteration of β-catenin expression in colonic epithelial cells of familial adenomatous polyposis patients. Cancer Res 56: 2213–2217.
Izban KF, Ergin M, Huang Q, Qin JZ, Martinez RL, Schnitzer B, Ni HY, Nickoloff BJ, and Alkan S (2001). Characterization of NF-κB expression in Hodgkin’s disease: Inhibition of constitutively expressed NF-κB results in spontaneous caspase-independent apoptosis in Hodgkin and Reed-Sternberg cells. Mod Pathol 14: 297–310.
Jones ME, Haire MF, Kloetzel PM, Mykles DL, and Schwartz LM (1995). Changes in the structure and function of the multicatalytic proteinase (proteasome) during programmed cell death in the intersegmental muscles of the hawkmoth, Manduca sexta. Dev Biol 169: 436–447.
Josefsberg LBY, Galiani D, Dantes A, Amsterdam A, and Dekel N (2000). The proteasome is involved in the first metaphase-to-anaphase transition of miosis in rat oocytes. Biol Reprod 62: 1270–1277.
Josefsberg LBY, Kaufmann O, Galiani D, Kovo M, and Dekel N (2001). Inactivation of M-phase promoting factor at exit from first embryonic mitosis in the rat is independent of cyclin B1 degradation. Biol Reprod 64: 871–878.
Kanayama H, Tanaka K, Aki M, Kagawa S, Miyaji H, Satoh M, Satoh M, Okada F, Sato S, Shimbara N, and Ichihara A (1991). Changes in expression of proteasome and ubiquitin genes in human renal cancer cells. Cancer Res 51: 6677–6685.
Katayose Y, Kim M, Rakkar ANS, Li Z, Cowan KH, and Seth P (1997). Promoting apoptosis: A novel activity associated with the cyclin-dependent kinase inhibitor p27. Cancer Res 57: 5441–5445.
Katayose Y, Wersto R, Cowan KH, and Seth P (1995). Effects of a recombinant adenovirus expressing WAF1/Cip1 on cell growth, cell cycle and apoptosis. Cell Growth Diff 6: 1207–1212.
Kawahara H, Kasahara M, Nishiyama A, Ohsumi K, Goto T, Kishimoto T, Saeki Y, Yokosawa H, Shimbara N, Murata S, Chiba T, Suzuki K, and Tanaka K (2000a). Developmentally regulated, alternative splicing of the Rpn10 gene generates multiple forms of 26S proteasomes. EMBO J 19: 4144–4153.
Kawahara H, Philipova R, Yokosawa H, Patel R, Tanaka K, and Whitaker M (2000b). Inhibiting proteasome activity causes overreplication of DNA and blocks entry into mitosis in sea urchin embryos. J Cell Sci 113: 2659–2670.
Kawana H, Tamaru J, Tanaka T, Hirai A, Saito Y, Kitagawa M, Mikata A, Harigaya K, and Kuriyama T (1998). Role of p27Kip1 and cyclin-dependent kinase 2 in the proliferation of non-small cell lung cancer. Am J Pathol 153: 505–513.
Keller JN, Hanni KB, and Markesbery WR (2000). Impaired proteasome function in Alzheimer’s disease. J Neurochem 75: 436–439.
Kitada T, Asakawa S, Matsumine H, Hattori N, Shimura H, Minoshima S, Shimizu M, and Mizuno Y (2000). Progress in the clinical and molecular genetics of familial parkinsonism. Neurogenet 2: 207–218.
Kitada T, Saskawa S, Hattori N, Matsumine H, Yanamura Y, Minoshima S, Yokoshi M, Mizuno Y, and Shimizu N (1998). Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392: 605–608.
Kitagawa H, Tani E, Ikemoto H, Ozaki I, Nakano A, and Omura S (1999). Proteasome inhibitors induce mitochondria-independent apoptosis in human glioma cells. FEBS Lett 443: 181–186.
Klein U, Gernold M, and Kloetzel PM (1990). Cell-specific accumulation of Drosophila proteasomes (MCP) during early embryonal development. J Cell Biol 111: 2275–2282.
Koepp DM, Harper WJ, and Elledge SJ (1999). How the cyclin became a cyclin: Regulated proteolysis in the cell cycle. Cell 97: 431–434.
Kordes U, Krappmann D, Heissmeyer V, Ludwig WD, and Scheidereit C (2000). Transcription factor NF-κB is constitutively activated in acute lymphoblastic leukemia cells. Leukemia 14: 399–402.
Korink V, Backer N, Morin PJ, van Wichen D, de Weger R, Kinzler KW, Vogelstein B, and Clevers H (1997). Constitutive transcriptional activation by a β-catenin-Tcf complex in APC−/− colon carcinoma. Science 275: 1784–1787.
Kudo Y, Takata T, Ogawa I, Kaneda T, Sato S, Takekoshi T, Zhao M, Miyauchi M, and Nikai H (2000). P27(Kip1) accumulation by inhibition of proteasome function induces apoptosis in oral squamous cell carcinoma cells. Clin Cancer Res 6: 916–923.
Kumamoto T, Fujimoto S, Ito T, Horinouchi H, Ueyama H, and Tsuda T (2000). Proteasome expression in the skeletal muscles of patients with muscular dystrophy. Acta Neuropathol 100: 595–602.
Kumatori A, Tanaka K, Inamura N, Sone S, Ogura T, Matsumoto T, Tachikawa T, Shin S, and Ichihara A (1990). Abnormally high expression of proteasomes in human leukemic cells. Proc Natl Acad Sci USA 87: 7071–7075.
Lam YA, Pickart CM, Alban A, Landon M, Jamieson C, Ramage R, Mayer RJ, and Layfield R (2000). Inhibition of the ubiquitin-proteasome system in Alzheimer’s disease. Proc Natl Acad Sci USA 97: 9902–9906.
Lee DH and Goldberg AL (1998). Proteasome inhibitors: Valuable new tools for cell biologists. Trends Cell Biol 8: 397–403.
Levine AJ (1997). p53, the cellular gatekeeper for growth and division. Cell 88: 323–331.
Li B and Dou P (2000). Bax degradation by the ubiquitin/proteasome-dependent pathway: Involvement in tumor survival and progression. Proc Natl Acad Sci USA 97: 3850–3855.
Lin L, DeMartino GN, and Greene WC (1998). Cotranslational biogenesis of NF-κB p50 by the 26S proteasome. Cell 92: 819–828.
Loda M, Cukor B, Tam SW, Lavin P, Fiorentino M, Draetta GF, Jessup JM, and Pagano M (1997). Increased proteasome-dependent degradation of the cyclin-dependent kinase inhibitor p27 in aggressive colorectal carcinomas. Nat Med 3: 231–234.
Lopes UG, Erhardt P, Yao R, and Cooper GM (1997). p53-dependent induction of apoptosis by proteasome inhibitors. J Biol Chem 272: 12893–12896.
Löw P, Bussell K, Dawson SP, Billett MA, Mayer RJ, and Reynolds SE (1997). Expression of a 26S proteasome ATP-ase subunit, MS73, in muscles that undergo developmentally programmed cell death, and its control by ecdysteroid hormones in the insect Manduca sexta. FEBS Lett 400: 345–349.
Löwe J, Stock D, Jap B, Zwickl P, Baumeister W, and Huber R (1995). Crystal structure of the 20S proteasome from the archeon T. acidophilum at 3.4 Å resolution. Science 268: 533–539.
Luo Y, Hurwitz J, and Massague J (1995). Cell cycle inhibition by independent CDK and PCNA binding domains in p21Cip1. Nature 375: 159–161.
Maki CG, Huibregtse JM, and Howley PM (1996). In vivo ubiquitination and proteasome-mediated degradation of p53. Cancer Res 56: 2649–2654.
Mann C and Hilt W (2000). The ubiquitin-proteasome sytem in cell cycle control. In: Hilt W, and Wolf DH, editors. Proteasomes: The world of regulatory proteolysis. Georgetown, Texas: Landes Bioscience, 264–301.
Marambaud P, Ancolio K, Lopez-Perez E, and Checler F (1998). Proteasome inhibitors prevent the degradation of familial Alzheimer’s disease-linked presenilin 1 and potentiate Aβ42 recovery from human cells. Mol Med 4: 147–157.
Marambaud P, Chevallier N, Barelli H, Wilk S, and Checler F (1997). Proteasome contributes to the α-secretase pathway of amyloid precursor protein in human cells. J Neurochem 68: 698–703.
Marti A, Wirbelauer C, Scheffner M, and Krek W (1999). Interaction between ubiquitin-protein ligase SCFSKP2 and E2F-1 underlies the regulation of E2F-1 degradation. Nat Cell Biol 1: 14–19.
Martin-Aparicio E, Yanamoto A, Hernandez F, Hen R, Avila JS, and Lucas JJ (2001). Proteasomal-dependent aggregate reversal and absence of cell death in a conditional mouse model of Huntington’s disease. J Neurosci 21: 8772–8781.
Masdehors P, Merle-Beral H, Maloum K, Omura S, Magdelenat H, and Delic J (2000). Deregulation of the ubiquitin system and p53 proteolysis modify the apoptotic response in B-CLL lymphocytes. Blood 96: 269–274.
Matilla A, Gorbea C, Einum DD, Townsend J, Michalik A, van Broeckhoven C, Jensen CC, Murphy KJ, Ptacek LJ, and Fu YH (2001). Association of ataxin-7 with the proteasome subunit S4 of the 19S regulatory complex. Hum Mol Genet 10: 2821–2831.
Mayo MW, Wang CY, Cogswell PC, Rogers Graham KS, Lowe SW, Der CJ, and Baldwin AS (1997). Requirement of NF-κB activation to suppress p53-independent apoptosis induced by oncogenic Ras. Science 278: 1812–1815.
McGarry TJ and Kirschner MW (1998). Geminin, an inhibitor of DNA replication, is degraded during mitosis. Cell 93: 1043–1053.
McGhee JD (1995). Cell fate decisions in the early embryo of the nematode Caenorhabditis elegans. Dev Genet 17: 155–166.
McNaught KSP and Jenner P (2001). Proteasomal function is impaired in substantia nigra in Parkinson’s disease. Neurosci Lett 297: 191–197.
Miyashita T and Reed JC (1995). Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 80: 293–299.
Monney L, Otter I, Olivier R, Ozer HL, Haas AL, Omura S, and Borner C (1998). Defects in the ubiquitin pathway induce caspase-independent apoptosis blocked by Bcl-2. J Biol Chem 273: 6121–6131.
Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, and Kinzler KW (1997). Activation of β-catenin Tcf-signaling in colon cancer by mutations in β-catenin and APC. Science 275: 1787–1790.
Morisato D and Anderson KV (1995). Signaling pathways that establish the dorsal-ventral pattern of the Drosophila embryo. Annu Rev Genet 29: 371–399.
Naujokat C, Sezer O, Zinke H, Leclere A, Hauptmann S, and Possinger K (2000). Proteasome inhibitors induce caspase-dependent apoptosis and accumulation of p21WAF1/Cip1 in human immature leukemic cells. Eur J Haematol 65: 221–236.
Navon A and Goldberg AL (2001). Proteins are unfolded on the surface of the ATPase ring before transport into the proteasome. Mol Cell 8: 1339–1349.
Negishi Y, Ui N, Nakajima M, Kawashima K, Maruyama K, Takizawa T, and Endo H (2001). p21Cip-1/SDI-1/WAF-1 gene is involved in chondrogenic differentiation of ATDC5 cells in vitro. J Biol Chem 276: 33249–33256.
Ni H, Ergin M, Huang Q, Qin JZ, Amin HM, Martinez RL, Saeed S, Barton K, and Alkan S (2001). Analysis of expresssion of nuclear factor kappa B (NF-κB) in multiple myeloma: Downregulation of NF-κB induces apoptosis. Br J Haematol 115: 279–286.
Ohtsubo M, Theodoras AM, Schumacher J, Roberts JM, and Pagano M (1995). Human cyclin E, a nuclear protein essential for the G1-to-S phase transition. Mol Cell Biol 15: 2612–2624.
Orlowski RZ (1999). The role of the ubiquitin-proteasome pathway in apoptosis. Cell Death Diff 6: 303–313.
Pagano M, Tam SW, Theodoras AM, Beer-Romero P, Del Sal G, Chau V, Yew PR, Draetta GF, and Rolfe M (1995). Role of the ubiquitin-proteasome pathway in regulating abundance of the cyclin-dependent kinase inhibitor p27. Science 269: 682–685.
Pahl HL and Baeuerle PA (1996). Control of gene expression by proteolysis. Curr Opin Cell Biol 8: 340–347.
Palombella VJ, Rando OJ, Goldberg AL, and Maniatis T (1994). The ubiquitin-proteasome pathway is required for processing the NF-kappa B1 precursor protein and the activation of NF-kappa B. Cell 78: 773–785.
Peterson BO, Wagener C, Marinoni F, Kramer ER, Melixetian M, Denchi EL, Gieffers C, Matteucci C, Peters JM, and Helin K (2000). Cell cycle- and cell growth-regulated proteolysis of mammalian Cdc6 is dependent on APC-Cdh1. Genes Dev 14: 2330–2343.
Piva R, Cancelli I, Cavalla P, Bortolotto S, Dominguez J, Draetta GF, and Schiffer D (1999). Proteasome-dependent degradation of p27/kip1 in gliomas. J Neuropathol Exp Neurol 58: 691–696.
Porter PL, Malone KE, Heagerty PJ, Alexander GM, Gatti LA, Firpo EJ, Daling JR, and Roberts JM (1997). Expression of cell cycle regulators p27Kip1 and cyclin E, alone or in combination, correlate with survival in young breast cancer patients. Nat Med 3: 222–225.
Powell SM, Zilz N, Beizer-Barclay Y, Bryan T, Hamilton S, Thibodean S, Vogelstein B, and Kinzler K (1992). APC mutations occur during colorectal tumorigenesis. Nature 359: 235–237.
Qiu JH, Asai A, Chi S, Saito N, Hamada H, and Kirino T (2000). Proteasome inhibitors induce cytochrome c-caspase-3-like protease-mediated apoptosis in cultured cortical neurons. J Neurosci 20: 259–265.
Reverte CG, Ahearn MD, and Hake LE (2001). CPEB degradation during oocyte maturation requires a PEST domain and the 26S proteasome. Dev Biol 231: 447–458.
Rideout HJ, Larsen KE, Sulzer D, and Stefanis L (2001). Proteasomal inhibition leads to formation of ubiquitin/α-synuclein-immunoreactive inclusions in PC12 cells. J Neurochem 78: 899–908.
Rieber M and Rieber MS (2000). Apoptosis-inducing levels of UV radiation and proteasome inhibitors produce opposite effects on p21WAF1 in human melanoma cells. Int J Cancer 86: 462–467.
Russel A, Thompson MA, Hendley J, Trute L, Armes J, and Germain D (1999). Cyclin D1 and D3 associate with the SCF complex and are coordinately elevated in breast cancer. Oncogene 18: 1983–1991.
Sadoul R, Fernandez PA, Quiquerez AL, Martinou I, Maki M, Schröter M, Becherer JD, Irmler M, Tschopp J, and Martinou JC (1996). Involvement of the proteasome in the programmed cell death of NGF-deprived sympathetic neurons. EMBO J 15: 3845–3852.
Saito Y and Kawashima S (1989). The neurite-initiation effect of a tripeptide aldehyde protease inhibitor on PC12h cells. J Biochem 106: 1035–1040.
Salon ML, Morelli L, Castano EM, Soto EF, and Pasquini JM (2000). Defective ubiquitination of cerebral proteins in Alzheimer’s disease. J Neurosci Res 62: 302–310.
Sawada MT, Morinaga C, Izumi K, and Sawada H (1999). The 26S assembly is regulated by a maturation-induced hormone in starfish oocytes. Biochem Biophys Res Commun 254: 338–344.
Scheffner M, Werness BA, Huibregtse JM, Levine AJ, and Howley PM (1990). The E6 oncoprotein encoded by human papilloma virus types 16 and 18 promotes the degradation of p53. Cell 63: 1129–1136.
Schreiber M, Müller WJ, Singh G, and Graham FL (1999). Comparison of the effectiveness of adenovirus vectors expressing cyclin kinase inhibitors p16INK4A, p18INK4B, p96INK4C, p21WAF1/Cip1, p27Kip1, in inducing cell cycle arrest, apoptosis and inhibition of tumorigenicity. Oncogene 18: 1663–1676.
Schwartz LM and Truman JW (1983). Hormonal control of rates of metamorphic development in the tobacco hornworm Manduca sexta. Dev Biol 99: 103–114.
Seeger M, Kraft R, Ferrell K, Bech-Otschir D, Dumdey R, Schade R, Gordon C, Naumann M, and Dubiel W (1998). A novel protein complex involved in signal transduction possessing similarities to 26S proteasome subunits. FASEB J 12: 469–478.
Sellin JH, Umar S, Xiao J, and Morris AP (2001). Increased β-catenin expression and nuclear translocation accompany cellular hyperproliferation in vivo. Cancer Res 61: 2899–2906.
Sgambato A, Cittadini A, Faraglia B, and Weinstein IB (2000). Multiple functions of p27Kip1 and its alteration in tumor cells: A review. J Cell Phisiol 183: 18–27.
Sheaff RJ, Singer JD, Swanger J, Smitherman M, Roberts JM, and Clurman BE (2000). Proteasomal turnover of p21Cip1 does not require p21Cip1 ubiquitination. Mol Cell 5: 403–410.
Sheikh MS, Rochefort H, and Garcia M (1995). Overexpression of p21WAF1/Cip1 induces growth arrest, giant cell formation and apoptosis in human breast carcinoma cell lines. Oncogene 11: 1899–1905.
Sherr CJ and Roberts JM (1999). CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev 13: 1501–1512.
Shimbara N, Orino E, Sone S, Ogura T, Takashina M, Shono M, Tamura T, Yasuda H, Tanaka K, and Ichihara A (1992). Regulation of gene expression of proteasomes (multi-protease complexes) during growth and differentiation of human hematopoietic cells. J Biol Chem 267: 18100–18109.
Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K, and Suzuki T (2000). Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet 25: 302–305.
Shinohara K, Tomioka M, Nakano H, Toné S, Ito H, and Kawashima S (1996). Apoptosis induction resulting from proteasome inhibition. Biochem J 317: 385–388.
Shirane M, Harumiya Y, Ishida N, Hirai A, Miyamoto C, Hatakeyama S, Nakayama K, and Kitagawa M (1999). Down-regulation of p27Kip1 by two mechanisms, ubiquitin-mediated degradation and proteolytic processing. J Biol Chem 274: 13886–13893.
Singer JD, Gurian-West M, Clurman B, and Roberts JM (1999). Cullin-3 targtes cyclin E for ubiquitination and controls S-phase in mammalian cells. Genes Dev 13: 2375–2387.
Sionov RV and Haupt Y (1999). The cellular response to p53: The decision between life and death. Oncogene 18: 6145–6157.
Slingerland J and Pagano M (2000). Regulation of the Cdk inhibitor p27 and its deregulation in cancer. J Cell Physiol 183: 10–17.
Spataro V, Norbury C, and Harris AL (1998). The ubiquitin-proteasome pathway in cancer. Brit J Cancer 77: 448–455.
Spinella MJ, Freemantle SJ, Sekula D, Chang JH, Christie AJ, and Dmitrovsky E (1999). Retinoid acid promotes ubiquitination and proteolysis of cyclin D1 during induced tumor cell differentiation. J Biol Chem 274: 22013–22018.
Stefanelli C, Bonavita F, Stanic I, Pignatti C, Farruggia G, Masotti L, Guarnieri C, and Caldarera CM (1998). Inhibition of etoposide-induced apoptosis with peptide aldehyde inhibitors of proteasome. Biochem J 332: 661–665.
Sutterluty H, Chatelain E, Marti A, Wirbelauaer C, Senften N, Müller U, and Krek W (1999). p45SKP2 promotes p27Kip1 degradation and induces S phase in quiescent cells. Nat Cell Biol 1: 207–214.
Suzuki Y, Nakabayashi Y, and Takahashi R (2001). Ubiquitin-protein ligase activity of X-linked inhibitor of apoptosis protein promotes proteasomal degradation of caspase-3 and enhances its anti-apoptotic effect in Fas-induced cell death. Proc Natl Acad Sci USA 98: 8662–8667.
Tamura N, Pfeifer G, Baumeister W, and Tamura T (2001). Tricorn protease in bacteria: Characterization of the enzyme from Streptomyces coelicor. Biol Chem 382: 449–458.
Tanaka K, Suzuki T, Chiba T, Shimura H, Hattori N, and Mizuno Y (2001a). Parkin is linked to the ubiquitin pathway. J Mol Med 79: 482–494.
Tanaka Y, Engelender S, Igarashi S, Rao RK, Wanner T, Tanzi RE, Sawa A, Dawson VL, Dawson TM, and Ross CA (2001b). Inducible expression of mutant α-synuclein decreases proteasome activity and increases sensitivity to mitochondria-dependent apoptosis. Hum Mol Genet 10: 919–926.
Tanimoto Y, Onishi Y, Hashimoto S, and Kizaki H (1997). Peptidyl aldehyde inhibitors of proteasome induce apoptosis rapidly in mouse lymphoma RVC cells. J Biochem 121: 542–549.
Tatebe H and Yanagida M (2000). Cut8, essential for anaphase, controls localization of 26S proteasome, facilitating destruction of cyclin and Cut2. Curr Biol 10: 1329–1338.
Thullberg M, Bartek J, and Lukas J (2000). Ubiquitin/proteasome-mediated degradation of p19(INK 4d) determines its periodic expression during the cell cycle. Oncogene 19: 2870–2876.
Tofaris GK, Layfield R, and Spillantini MG (2001). α-synuclein metabolism and aggregation is linked to ubiquitin-independent degradation by the proteasome. FEBS Lett 509: 22–26.
Tokumoto T, Tokumoto M, Seto K, Horiguchi R, Nagahama Y, Yamada S, Ishikawa K, and Lohka MJ (1999). Disappearance of a novel protein component of the 26S proteasome during Xenopous oocyte maturation. Exp Cell Res 247: 313–319.
Tomoda K, Kubota Y, and Kato J (1999). Degradation of the cyclin-dependent-kinase inhibitor p27Kip1 is instigated by Jab1. Nature 398: 160–165.
Tsubuki S, Kawasaki H, Saito Y, Miyashita N, Inomata M, and Kawashima S (1993). Purification and characterization of a Z-Leu-Leu-Leu-MCA degrading protease expected to regulate neurite formation: A novel catalytic activity in proteasome. Biochem Biophys Res Commun 196: 1195–1201.
Tsubuki S, Saito Y, Tomioka M, Ito H, and Kawashima S (1996). Differential Inhibition of calpain and proteasome activities by aldehydes of di-leucine and tri-leucine. J Biochem 119: 572–576.
Tyers M and Jorgensen P (2000). Proteolysis and the cell cycle: With this RING I do thee destroy. Curr Opin Genet Dev 10: 54–64.
Urano T, Yashiroda H, Muraoka M, Tanaka K, Hosoi T, Inoue S, Ouchi Y, Tanaka K, and Toyoshima H (1999). p57Kip2 is degraded through the proteasome in osteoblasts stimulated to proliferation by transforming growth factor β1. J Biol Chem 274: 12197–12200.
Verma IM, Stevenson JK, Schwartz EM, van Antwerp D, and Miyamoto S (1995). Rel/NK-κB/IκB family: Intimate tales of association and dissociation. Genes Dev 9: 2723–2735.
Voges D, Zwickl P, and Baumeister W (1999). The 26S proteasome: A molecular machine designed for controlled proteolysis. Annu Rev Biochem 68: 1015–1068.
Wang CY, Mayo MW, Korneluk RG, Goeddel DV, and Balwin AS (1998). NF-kappaB antiapoptosis: Induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science 281: 1680–1683.
Wang QM, Jones JB, and Studzinski GP (1996). Cyclin-dependent kinase inhibitor p27 as a mediator of the G1-S phase block induced by 1,25-dihydroxyvitamin D3 in HL-60 cells. Cancer Res 56: 264–267.
Wang X, Gorospe M, Huang Y, and Holbrook NJ (1997). p27Kip1 overexpression causes apoptotic death of mammalian cells. Oncogene 15: 2991–2997.
Wei N, Tsuge T, Serino G, Dohmae N, Takio K, Matsui M, and Deng XW (1998). The COP9 complex is conserved between plants and mammals and is related to the 26S proteasome regulatory complex. Curr Biol 8: 919–922.
Williams A, Sun XY, Fischer JE, and Hasselgren PO (1999). The expression of genes in the ubiquitin-proteasome proteolytic pathway is increased in skeletal muscle from patients with cancer. Surgery 126: 744–749.
Winston JT, Chu C, and Harper JW (1999). Culprits in the degradation of cyclin E apprehended. Genes Dev 13: 2751–2757.
Xie Y and Varshavsky A (2000). Physical association of ubiquitin ligases and the 26S proteasome. Proc Natl Acad Sci USA 97: 2497–2502.
Yang Y, Fang S, Jensen JP, Weissman A, and Ashwell JD (2000). Ubiquitin protein ligase of IAPs and their degradation in proteasomes in response to apoptotic stimuli. Science 288: 874–877.
Yew PR (2001). Ubiquitin-mediated proteolysis of vertebrate G1- and S-phase regulators. J Cell Physiol 187: 1–10.
Zavitz KH and Zipursky SL (1997). Controlling cell proliferation in differentiation: Genetic analysis of negative regulators of G1- → S-phase progression. Curr Opin Cell Biol 9: 773–781.
Zhang J, Houston DW, King ML, Payne C, Wylie C, and Heasman J (1998). The role of maternal VegT in establishing the primary germ layers in Xenopus embryos. Cell 94: 515–524.
Acknowledgements
We are grateful to all of the colleagues who have given critical comments on this work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Naujokat, C., Hoffmann, S. Role and Function of the 26S Proteasome in Proliferation and Apoptosis. Lab Invest 82, 965–980 (2002). https://doi.org/10.1097/01.LAB.0000022226.23741.37
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1097/01.LAB.0000022226.23741.37
This article is cited by
-
Tachykinin-related peptides modulate immune-gene expression in the mealworm beetle Tenebrio molitor L.
Scientific Reports (2022)
-
Marine-derived pipeline anticancer natural products: a review of their pharmacotherapeutic potential and molecular mechanisms
Future Journal of Pharmaceutical Sciences (2021)
-
Simultaneous Disruption of Both Polyubiquitin Genes Affects Proteasome Function and Decreases Cellular Proliferation
Cell Biochemistry and Biophysics (2020)
-
A new pyridazinone exhibits potent cytotoxicity on human cancer cells via apoptosis and poly-ubiquitinated protein accumulation
Cell Biology and Toxicology (2019)
-
Association between functional variant of inflammatory system gene (PSMA6) and end-stage kidney disease
International Urology and Nephrology (2016)
