
Abstract
Only a few studies have demonstrated successful molecular analysis after whole genome amplification using single cells dissected from paraffin-embedded tissues. The results in these studies were limited by low-amplification efficiency and high rates of allele dropout. In the present study, the amplification rate using a thoroughly modified primer extension and preamplification-PCR protocol was improved significantly for single cells microdissected from paraffin-embedded and immunohistochemically stained tissues. Tissue fixation with ethanol (85%) and the addition of 0.2 mmol/L EDTA helped to achieve an amplification rate between 67% (segments 200 to 400 bp) and 72% (segments <200 bp). Normal tissue sections were immunohistochemically double stained for overabundance of p53 protein and proliferating cell nuclear antigen. Microdissection of single cells was performed with a manual micromanipulator equipped with a Tungsten needle. Sequence analysis of the TP53 gene was performed after improved primer extension preamplification-PCR and multiplex PCR from single microdissected cells. The rate of allele dropout was at least 68%. These technical advances facilitate routine mutation analysis using a single cell or a few cells microdissected from routinely processed paraffin-embedded normal and tumor tissues. Allele dropout still represents a serious problem in single-cell mutation analysis, especially in samples with limited template DNA and prone to DNA damage.
Similar content being viewed by others
Introduction
Molecular analysis of single cells has become routine in reproductive medicine (Hahn et al, 2000). However, few studies have routinely analyzed rare heterozygous mutations in single cells microdissected from fixed, paraffin-embedded, and immunohistochemically stained tissue sections because of certain limitations (Becker et al, 1996; Roehrl et al, 1997; Schutze and Lahr, 1998; Suarez-Quian et al, 1999). Crosslinking of DNA in tissues fixed with the commonly used fixative, formalin, limits the efficiency of PCR amplification and reduces the maximum length of the PCR amplicon (Karlsen et al, 1994; Williams et al, 1999). Allele dropout (ADO) occurs when one allele is preferentially amplified over the other to the extent that the minor allele is not detected. ADO is particularly problematic when templates consisting of DNA from less than 10 cells are amplified; it occurs at the same rate irrespective of whether nested PCR or whole genome amplification (WGA) is used for initial amplification (Hahn et al, 1998, 2000). ADO is especially problematic in the diagnosis of rare heterozygous mutations from single cells. No protocol has been identified that eliminates ADO (Hahn et al, 2000). Of great interest is the development of methodologies that permit examination of rare heterozygous mutations in single cells from normal tissues with high sensitivity and accuracy to enable investigation of the role of mutagenesis and risk of disease.
Immunohistochemistry is a technique that specifically provides information about protein expression patterns at the single-cell level and may indicate molecular changes relevant to neoplastic development. The tumor suppressor gene TP53 is mutated in 50% of human cancers (Greenblatt et al, 1994). In its mutated form or under certain cellular and/or genotoxic stress, the p53 protein becomes stabilized and its overabundance is readily detectable by immunohistochemical staining (May and May, 1999; Wallace-Brodeur and Lowe, 1999). Overabundance of p53 protein may be found even in normal tissues in the nuclei of cells with a morphologically normal appearance (Barnes et al, 1992; Huusko et al, 1999; Varley et al, 1999). Immunohistochemical staining for p53 can be used to identify cells that are potential candidates for TP53 gene mutations. Because proliferating cell nuclear antigen (PCNA) expression is regulated by p53, double immunostaining for overexpression of p53 and PCNA proteins was used to enhance detection of cells with overexpression of p53 caused by a mutation in the p53 gene of functional relevance.
In general, for molecular analyses from single cells, two different PCR techniques can be used: direct (usually nested) amplification or (nonspecific) preamplification of the genetic material by WGA followed by specific (nested) PCR. A major advantage of WGA is the possibility of multiple genetic analyses from a single cell. Of the various WGA techniques available, primer extension and preamplification (PEP)-PCR and degenerate oligonucleotide-primed PCR provide the most complete coverage of the genome (Wells et al, 1999). Improved primer extension and preamplification (I-PEP)-PCR is superior to degenerate oligonucleotide-primed PCR in amplification efficiency (Dietmaier et al, 1999). PEP-PCR is a technique of WGA used before specific PCR from minute amounts of DNA, which enables the molecular analysis of multiple genes from a single cell or a few cells (Zhang et al, 1992). PEP-PCR has recently been improved to facilitate multiple genetic analyses of small cell clusters microdissected from formalin-fixed paraffin-embedded tissues (Dietmaier et al, 1999). Although some studies have analyzed DNA from immunohistochemically stained single cells from fresh-frozen tissue samples (Persson et al, 2000; Ponten et al, 1997), to our knowledge, no study has conducted WGA before specific amplification of single-cell genomes from fixed, paraffin-embedded, and immunohistochemically stained tissue sections. Herein, DNA integrity, amplification efficiency, and ADO are evaluated after an overall enhanced protocol for microdissection of single cells from ethanol-fixed, paraffin-embedded, and immunohistochemically stained tissue sections and amplification with a modified I-PEP-PCR protocol.
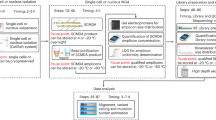
In this study major improvements to tissue fixation and processing and single-cell DNA amplification were made to enable the analysis of rare heterozygous mutations in the TP53 gene of single cells microdissected from ethanol-fixed, paraffin-embedded, and immunohistochemically stained sections of normal human tissue (Fig. 1).

A flow chart of the methods used for mutation analysis using ethanol-fixed, paraffin-embedded, and immunohistochemically stained single cells or small cell clusters.
Results
Tissue Fixation and Immunohistochemistry
To avoid DNA crosslinking by formalin fixation of tissue, which limits molecular analysis of single cells, ethanol/EDTA fixation was optimized to maintain high DNA integrity and provide high-resolution immunohistochemistry. Ethanol has a precipitating effect on tissues. EDTA inhibits DNases present in surgically removed tissues for better preservation of DNA. Proteinase K digestion of various tissues fixed with ethanol/EDTA (“Materials and Methods”) revealed the presence of single-stranded, high molecular weight DNA (∼20 Kb) after the final step of immunohistochemical staining in all tissues analyzed (data not shown). To demonstrate the feasibility of ethanol/EDTA tissue fixation for immunohistochemistry, double staining of p53 protein (red-colored nuclear stain) together with PCNA (brown-colored nuclear stain) was used. A mix of 85% ethanol/0.2 mmol/L EDTA was found to give crisp and clear immunohistochemistry after a brief antigen retrieval by steam treatment at 95° C for 5 minutes, enabling the clear distinction of nuclear staining for p53 protein, or PCNA, or both p53 and PCNA in various tissues with physiologically different expression patterns of both proteins (Fig. 2). Fixation with 95% ethanol/0.1 mmol/L EDTA or 75% ethanol/1 mmol/L ETDA resulted in less clear staining (not shown). In addition, cutting of sections was impaired when 95% ethanol/0.1 mmol/L EDTA was used as fixative.

Immunohistochemical staining for p53 and PCNA protein (overabundance) in nucleus and cytoplasm, respectively, for normal human tissues that have been fixed in ethanol and embedded in paraffin. A, Normal colon tissue with crypt epithelial cells staining positive for PCNA (brown cytoplasmic stain) and a single cell staining positive (red nuclear stain) for p53 overabundance (arrow) (magnification ×100). B, Normal lung tissue with bronchial epithelial cells. A cell staining for PCNA (brown cytoplasmic stain, white arrow) can be seen in addition to a cell staining for p53 overabundance (red nuclear stain, black arrow) (magnification ×40). C, Benign proliferative lesion of the mammary gland. A cell staining red for p53 protein overabundance (white arrow) is seen together with a cell staining mixed red-brown for both p53 and PCNA (black arrow) proteins and many other cells staining brown for PCNA overabundance (magnification ×250).
Optimization of I-PEP-PCR: Analysis of Dilutions of Genomic DNA
Preliminary experiments indicated that the amplification success rate of various exons of the TP53 gene from single cells microdissected from ethanol/EDTA-fixed tissues was 30% or less when the I-PEP technique originally described by Dietmaier et al (1999) was used. Such a low amplification success rate from single cells would make the routine analysis of genes like TP53 costly and cumbersome, because exons 5 to 9, which are commonly examined, would be amplified in only a small number of microdissected single cells. To optimize I-PEP, genomic DNA was serially diluted to the single-cell level (6 pg). To ensure optimal digestion, the concentration of Tween-20 in the digestion buffer was increased from 0.5% to 3%. To achieve maximum amplification efficiency, various PCR cycling conditions (annealing temperature, extension time) and I-PEP-PCR mix ingredients (concentration of primers, MgCl2, dNTP, enzymes) were tested. The concentrations of MgCl2 and dNTP were increased stepwise from 2.5 mmol/L and 0.1 mmol/L to 6.0 mmol/L and 1 mmol/L, respectively. Specific PCR after I-PEP-PCR was most effective when MgCl2 was 6.0 mmol/L and dNTPs were 1 mmol/L each in I-PEP-PCR. Of the primer concentrations tested (5 μmol/L, 10 μmol/L, 15 μmol/L, 20 μmol/L, 25 μmol/L, 30 μmol/L), 20 μmol/L was found to be optimal. Of the various enzymes tested (Taq Expand High Fidelity Polymerase with and without Bst DNA polymerase [Roche, Basel, Switzerland], Pfu Turbo [Stratagene, La Jolla, California], and AdvanTaq [Clontech Laboratories, Palo Alto, California]), Taq Expand High Fidelity Polymerase without Bst DNA polymerase was found to be superior. No amplification was seen when Pfu Turbo was used in I-PEP (not shown). Lowering the annealing temperature from 37° C to 28° C, reducing the annealing time from 4 minutes to 2 minutes, and prolonging the extension time from 30 seconds to 3 minutes enabled the amplification of exons 5 to 9 of the TP53 gene after I-PEP-PCR. A nested amplification of ∼2 Kb from high molecular weight genomic DNA was possible even to a dilution of 13.2 pg of template DNA, which is equivalent to four copies of mammalian genome.
Measurement of ADO After Amplification from Single Human Fibroblasts
Amplification efficiency using single fibroblasts (HF57 human fibroblasts) was assessed using amplification of various segment lengths within exons 5 to 9 of the TP53 gene in a nested PCR configuration after I-PEP. Unfixed fibroblasts were air dried for 2 hours and used for analysis of amplification from single cells and for measurement of ADO. Segments of ∼500 bp (eg, spanning exons 5 and 6) were amplified with a success rate of 68% (33 of 48 single cells); segments of 300 bp or less (eg, exon 6) were amplified with a success rate of 87% (42 of 48 single cells; not shown). A ∼2 Kb segment (spanning exons 5 to 9) could be amplified in 4 (8%) of 50 single cells after I-PEP.
HF57 human fibroblasts are heterozygous for a polymorphism (G>A bp 13494) in intron 6 of the TP53 gene, which makes them useful for the study of ADO after I-PEP-PCR from single cells. Exon 6 was amplified after I-PEP-PCR from 37 air-dried, single HF57 fibroblast cells. ADO was determined after DNA sequencing. The ADO rate was 68% (25 of 37 single cells). Twelve single cells (32%) demonstrated heterozygosity at bp 13494. Of the single cells with ADO, 32% (12 single cells) showed the mutant allele sequence G>A bp 13494, and 35% (13 single cells) exhibited the wild-type sequence (Fig. 3). ADO was not observed when five or more cells were used for I-PEP-PCR.

Evaluation of allele dropout (ADO) after improved primer extension and preamplification (I-PEP)-PCR. Sequence analysis of a heterozygote sequence change intron 6 bp 13494 (G>A) of TP53 gene in single HF57 fibroblasts. Lanes 1 and 3: single cell, heterozygosity; lanes 2, 4, and 5: single cell, wild-type, allele dropout; lane 6: single cell, G>A, ADO.
Amplification from Single Cells Microdissected from Ethanol-Fixed, Paraffin-Embedded, and Immunohistochemically Stained Tissue Sections
Amplification efficiency using single microdissected cells from ethanol-fixed, paraffin-embedded, and immunohistochemically stained tissue sections was determined by amplification of various exons of the TP53 gene after I-PEP-PCR and a nested PCR configuration. First-round amplification was performed in a multiplex PCR using primers covering exons 5 to 9 of the TP53 gene. Segments between 200 and 400 bp (single exons of TP53 gene) were amplified in up to 67% of cells; segments smaller than 200 bp were amplified in up to 72% of cells (Fig. 4). Amplification efficiency was dependent on the type of tissue analyzed. Pancreatic tissue has a high degree of enzymatic DNA degradation starting early during surgical manipulation. DNA amplification from pancreatic tissue was compared with colon tissue, in which DNA is usually not affected by enzymatic damage during surgery. As expected, amplification success was greater using single cells from colon than from pancreatic tissue. In a series of amplification experiments, all five exons (5 to 9) of the TP53 gene were amplified in 50% of single cells harvested from colon versus 36% from pancreatic tissue. The amplification rates of at least two exons were considerably higher in colon tissues (Table 3). High multiplex amplification success rate was achieved using tagged primers (see “Materials and Method”) in first-round PCR in addition to an antibody that ensures a hot start PCR by neutralizing Taq polymerase.

Representative examples of the amplification rate of various segments of exons 5 to 9 from colon cells after I-PEP-PCR. Upper: Nested PCR exon 9 (first-round segment 378 bp); middle: different samples with nested PCR exon 5 (first-round segment 383 bp); lower: different samples with nested PCR exon 5 (first-round segment 181 bp). Lane 1: 30 cells; lanes 2 to 16: single cell; lanes 17 and 18: negative control, no cells.
Identification of Rare Heterozygous Mutations in Single Cells of Normal Colon Identified To Have an Overabundance of P53 Protein
Normal colon tissue from five patients who were treated for colon carcinoma was fixed in ethanol/EDTA, embedded in paraffin, and immunohistochemically stained for identification of p53 protein overexpression. Normal tissues from two of these patients did not stain positive for p53 protein. Normal tissues from the remaining three patients exhibited a positive nuclear staining for p53 protein in up to nine epithelial cells/slide; the total number of epithelial cells per slide was estimated to be between 40,000 and 60,000 cells. A total of 23 single cells and a cell cluster (40 cells) with positive p53 protein immunohistochemistry were microdissected and amplified. Sequence analysis revealed five sequence changes (Fig.5).

Identification of mutations in exons 5 to 9 of the TP53 gene from single cells that were immunohistochemically stained positive for p53 protein overabundance. Please note that arrows are used to mark the mutant sequence. The single cells were from paraffin-embedded, normal colon tissue. A, Forty cells with a heterozygous mutation: C to T transition (arrow) exon 7 bp 14,049 (pro/val). B, Single cell with deletion of four bases (ATGG) and insertion of three bases (“TAA” arrows) exon 5 (bp 13,157 to 13,161). C, Single cell with a silent G to A transition (arrow) in exon 8 (bp 14,506). D, Single cell with a C to T transition (arrow) in exon 5 (bp 13,234, asp/val). E, Single cell with a heterozygous sequence change in intron 5 (G to A transition, bp 13,292).
Discussion
Formalin Fixation Limits Molecular Analysis, Especially Using Single Cells
There is no better place to study the underlying molecular genetic dynamics of normal and pathologic processes than in vivo. Histologic tissue sections are one way to gain access to a fingerprint of these processes within a defined morphologic compartment and at the level of a single cell (Emmert-Buck et al, 1996; Schutze and Lahr, 1998). However, technical limitations have hindered widespread genomic analyses especially at the single-cell level in large part because of the routine formalin fixation of tissues, which causes DNA crosslinking, thereby reducing PCR efficiency. This is one of the reasons why the achieved amplification rates reported from single cells microdissected from formalin-fixed paraffin-embedded tissue slides did not exceed 35% in short segments up to 246 bp (Becker et al, 1996; Roehrl et al, 1997). Considerably higher amplification rates (up to 70%) have been reported from single cells dissected from frozen, immunohistochemically stained tissues (Persson et al, 2000). However, major limitations in the analysis of frozen tissue sections include the usually considerably lower quality of immunohistochemical staining, especially if double staining is performed. In addition, the availability of frozen tissues for molecular analyses is usually much lower in comparison to paraffin-embedded tissues. To circumvent the disadvantages associated with formalin fixation and frozen tissues, tissues were fixed in an ethanol/EDTA solution before paraffin embedding.
Ethanol/EDTA Fixation Improves Molecular Analyses Using Single Cells
In the present study, a mixture of 85% ethanol/0.2 mmol/L EDTA gave crisp and clear immunohistochemical staining for two proteins (p53 and PCNA) simultaneously in various tissues, providing the basis for coupled morphologic and molecular analyses in paraffin-embedded tissues at a single cell-level. It has been demonstrated that ethanol fixation is the method of choice for immunohistochemical staining of a variety of tissue antigens, especially the p53 protein (Arnold et al, 1996; Bassarova and Popov, 1998). Furthermore, addition of EDTA has been shown to inhibit DNA degradation in formalin-fixed tissues to a certain degree (Yagi et al, 1996). The specificity of the immunohistochemical double staining for both proteins was demonstrated by frequent expression of both proteins in highly dividing tissues like colon and a proliferative lesion of the mammary gland, by moderate expression of both proteins in moderately dividing tissues like bronchial epithelium, and by rare expression of one of the proteins in slowly dividing tissues like liver tissue.
Modified I-PEP Shows Improved Amplification Success
Herein, a recently published I-PEP-PCR technique (Dietmaier et al, 1999) was thoroughly optimized. The revised protocol demonstrated amplification of ∼2 Kb segments after I-PEP-PCR when high molecular weight DNA was used as the template, even using dilutions down to 12 pg, ie, four copies (or two cells) of mammalian genome. Successful amplification of 2 Kb segments after PEP has been described previously but only after using 5000 cells as initial template (Duddy et al, 1998). Using air-dried single fibroblasts, a 2 Kb segment could be amplified but only with a small number of cells. This may be a result of the use of unfixed cells that were air dried for at least 2 hours, when oxygen-related damage may have already led to DNA degradation. When smaller segments were amplified after I-PEP-PCR, amplification efficiency was up to 87%. Most importantly, the amplification rate of segments covering single exons of the TP53 gene was increased from 30% to up to 72% from single ethanol-fixed, paraffin-embedded, and immunohistochemically stained single cells after the present modifications of I-PEP-PCR. As a result of this improvement, serial molecular analyses of single cells stained for overabundance of proteins like p53 or others can be done in a reasonable number of PCR experiments. Exons 5 to 9 of the TP53 gene were amplified in 50% of single cells dissected from colon samples. The amplification success rate in single pancreatic cells was lower, most likely because of the much higher degree of pancreatic protease-related DNA damage (autodigestion) beginning early during surgical manipulation of this tissue, in addition to the long surgical procedure if performed as Whipple procedure. But even under these most unfavorable conditions, multiplex PCR after WGA offers the opportunity of multiple genetic analyses of various genes even if multiple exons are to be analyzed. In addition, if mutations are found by sequence analysis, reamplification for confirmation of a particular base change can be performed from the original template, which is not possible when single cells are amplified directly by specific PCR without prior preamplification by any WGA method. Of interest is the rate of polymerase errors during the amplification from single cells. Herein, over 10,000 nucleotides were sequenced in the analysis of single, air-dried fibroblasts and not a single artificial mutation was observed.
ADO After Modified I-PEP Amplification from Single Cells Is Still High
ADO or preferential amplification of one allele over the other during PCR amplification is a well-known phenomenon limiting detection of heterozygote base changes (Hahn et al, 1998, 2000). ADO rates between 0% and 83% have been reported and are dependent on the source and quality of the template DNA amplified (Findlay et al, 1995, 1998; Garvin et al, 1998; Hahn et al, 1998; Rechitsky et al, 1996; Snabes et al, 1994; Wells et al, 1999). As a result of ADO, mutation analysis may fail to detect heterozygous mutations in a significant number of cases. The distribution of alleles dropping out during PCR amplification occurs at random. The occurrence of ADO is influenced by multiple factors, eg, cell lysis, conditions of PCR mix, thermal cycling conditions, guanine/cytosine content of the DNA segment amplified, or degradation of the DNA template (El-Hashemite and Delhanty, 1997; Ray and Handyside, 1996; Walsh et al, 1992). Furthermore, ADO occurs in either direct, specific (nested) amplification or in PEP-PCR before specific PCR from single cells (Hahn et al, 1998). This ADO estimate is specifically for the amplification method and is a minimal estimate because it is expected that ADO is much higher for analysis of single cells microdissected from paraffin-embedded tissues. No method described to date has been able to eliminate ADO.
Forty-one cell culture human fibroblasts harboring a heterozygous polymorphism in intron 6 of the TP53 gene were used for the analysis of ADO using the modified I-PEP-PCR protocol. Fibroblasts were spread on slides, air dried, and stained with methylene blue. Because fibroblasts were not prepared with a fixative like ethanol or acetone to ensure at least a limited protection, the fibroblasts were not protected from oxygen radical–induced DNA damage. Because DNA degradation starts immediately after cells are exposed to oxygen, this situation closely resembles the exposure of immunohistochemically stained cells to DNA damaging oxygen radicals while sitting as a single-cell layer on slides in the course of tissue processing and microdissection. Herein the ADO rate was 68% and affected both alleles randomly. If a heterozygous mutation is present, according to the data herein, the chance of the correct diagnosis of heterozygosity will be 22%.
Mutation Analysis in Single Cells from Ethanol-Fixed, Paraffin-Embedded, and Immunohistochemically Stained Normal Tissues
This is the first report of specific molecular analyses after WGA from immunohistochemically stained single cells microdissected from paraffin-embedded tissues. In the analysis of a limited number of microdissected single cells from normal colon tissue staining positive for p53 protein, sequence changes in the TP53 gene were found in 4 of 23 single cells and in 1 cell cluster of 40 cells. The sequence changes were not published mutational hotspots of the TP53 gene. The relevance of the heterozygote mutations detected in intron 5 of one single cell and the heterozygous base change seen in the cell cluster in regard to inactivation of the TP53 gene is not clear. However, heterozygous mutations inactivating the remaining wild-type allele have been described (Forrester et al, 1995; Hachiya et al, 1994; Milner and Medcalf, 1991; Villadsen et al, 2000). Possible reasons besides ADO for detecting only wild-type sequence in a subset of single cells with positive p53 immunohistochemistry dissected from tissue slides may be contamination of the PCR or loss of genetic material containing the allele harboring the base change during tissue cutting. The chance of losing genetic material from a single cell nucleus in the course of tissue sectioning can be estimated to be between 10% and 20% if sections of 6 μm are processed. It should be noted that switching from the analysis of single paraffin-embedded cells to single cells from fresh-frozen tissues might not be helpful in solving the ADO problem. A recently published article reported an ADO rate of 50% from single cells dissected from immunohistochemically stained fresh-frozen tissue sections (Persson et al, 2000).
Conclusion
In summary, we present a reproducible and reliable fixation protocol for paraffin-embedded tissues that allows clear and crisp immunohistochemical staining of at least two proteins simultaneously and preserves DNA integrity to a degree that allows the molecular analysis of multiple genes at the single-cell level. Single-cell microdissection was achieved by a fast and easy-to-handle simple manual microdissection device that is affordable to many laboratories, being about one tenth the price of a laser microdissection device. Furthermore, the success rate of PCR amplification from DNA obtained from a single cell dissected from paraffin-embedded tissue is increased to a level where serial analyses of mutations in a single cell or a few cells from normal or tumor tissue can be performed in a reasonable number of experiments. This could be the basis for further studies to determine an individual TP53 mutation load in normal tissues in the context of morphology, enabling an estimation of individual cancer risk well in advance of the development of malignant disease.
Material and Methods
Cell and Tissue Samples
Tissue samples from patients in surgery for colorectal cancer or pancreatic cancer were obtained from the Department of Pathology from the City of Hope National Medical Center (Duarte, California). Suspensions of cultured human fibroblasts (HF57) were a generous gift from Steven Bates (Department of Biology, City of Hope).
Tissue Processing and Immunohistochemistry
After tissues were handled for pathologic-anatomical diagnosis, samples were fixed immediately in 85% ethanol supplemented with 0.2 mmol/L EDTA for 24 hours and embedded in paraffin thereafter. Six-micron serial sections of ethanol (EDTA)-fixed, paraffin-embedded tissues were mounted on Probe-On slides (Ventana Medical Systems, Tucson, Arizona) and baked at 56° C for 1 hour. Slides were deparaffinized by incubating the slides in xylene for 2 × 10 minutes and rehydrating in 100% ethanol for 2 × 5 minutes, in 96% ethanol for 2 × 5 minutes, and in 70% ethanol for 2 × 5 minutes. Antigen retrieval was performed by placing the slides into steam heat for 5 minutes. Antibodies used for double staining were PCNA (Ab-1) (Oncogene Research Products, Cambridge, Massachusetts) and NCL-p53-DO7 (Novocastra, Newcastle-upon-Tyne, United Kingdom). Staining was performed using a Biotech Techmate 1000 Immunostainer (Ventana Medical Systems). Double staining of slides was performed using a modified ABC-Method (Ventana Medical Systems). Human PCNA was stained brown using the chromogen 3′3-diaminobenzidine, and the p53 protein was stained red using the chromogen Bio-Red. Appropriate positive tissue controls and negative controls for primary antibodies were included in every experiment. Tissue sections from a lung adenocarcinoma exhibiting p53 protein overexpression of tumor cells served as positive control. Staining without primary antibody was performed on the lung cancer tissue sections for demonstration of specificity of staining. Stained tissue sections were covered with aqueous mounting medium (Aquatex; Merck, Darmstadt, Germany) and a glass coverslip. Samples were stored frozen at −20° C until examined.
For determination of optimal tissue fixation and immunohistochemistry, fresh tissue samples were cut in three pieces and fixed in 95% ethanol/0.01 mmol/L EDTA, 85% ethanol/0.2 mmol/L EDTA, and 70% ethanol/1 mmol/L EDTA, respectively. After 24 hours, tissues were paraffin embedded, and immunohistochemical staining was performed as described above. The quality and reproducibility of staining was assessed independently by two pathologists (YS and LMW). DNA integrity was routinely assessed after Proteinase K digestion (2 mg/ml final concentration for 16 hours followed by a 10-minute inactivation step at 90° C) of cells from a variety of tissues scraped from serial sections taken at different stages of tissue processing and immunohistochemical staining. Eight-microliter aliquots of digested cell samples were electrophoresed through a 1% agarose gel for 30 minutes and stained with ethidium bromide.
Preparation of Single Cells from Cell Cultures and from Paraffin-Embedded, Immunohistochemically Stained Tissue Sections
Fibroblasts resuspended in PBS were spread onto glass slides and were air dried for 2 hours. After staining with methylene blue, single cells were picked with sterile needles (Microlance; Becton Dickinson, Franklin Lakes, New Jersey). Single cells or small cell clusters on tissue slides staining positive for p53 protein overabundance were microdissected using a manual micromanipulator (MP285; Sutter Instruments, Novato, California) equipped with a Tungsten needle (FHC Inc., Bowdoinham, Massachusetts). An inverted microscope (Nikon TMS, Melville, New York) was used. Microdissection was performed as follows: with a fine surgical needle and at lower magnification, the target cell was chosen and the area around it was freed from surrounding tissue by scraping a free space as close as possible to the target. After changing to a magnification of ×40, the cell was then pushed into the free space with the joystick-operated micromanipulator, picked up with a new surgical needle, and transferred into the PCR tube. Approximately 5 to 10 minutes are necessary to microdissect a single cell from normal colonic epithelium. Fibroblasts in a single-cell layer were dissected in less than 2 minutes per cell and could be dissected manually without the micromanipulator. Cells were transferred into 10 μl of 1× Taq PCR buffer No. 3 without MgCL2 (Roche) containing 400 μg/ml (fibroblasts) or 1 mg/ml (single cells from tissue sections) of Proteinase K and 3% Tween-20 (Roche). Cell lysis was performed by incubation for 3 hours (fibroblasts) or 16 hours (single cells from tissue sections) at 50° C followed by a 10-minute inactivation at 90° C.
PCR
A recently described protocol for improved primer extension preamplification (I-PEP)-PCR (Dietmaier et al, 1999) was used with major modifications (Table 1) in a Perkin Elmer 9600 thermocycler (Foster City, California). I-PEP-PCR was set up by adding 30 μl of I-PEP mix [final concentration: 0.05 mg/ml gelatin, 20 μmol/L (N)15 random primer, 1 mmol/L each dNTP, 3 U of Taq Expand High Fidelity polymerase, 6 mmol/L MgCl2, in 1× PCR buffer No. 3 from Roche] to 10 μl of lysis buffer containing the lysed cell. PCR was run for 50 cycles as follows: Step 1: 95° C for 2 minutes; Step 2: 95° C for 30 seconds; Step 3: 28° C for 90 seconds; Step 4: ramp 0.1° C per second to 55° C; Step 5: 55° C for 2 minutes; Step 6: 68° C for 3 minutes; Step 7: go to Step 2, 49 times; Step 8: 68° C for 15 minutes; and Step 9: 4° C. Exons 5 to 9 of the TP53 gene were amplified in a nested PCR approach: first-round PCR was performed as a multiplex PCR using upstream and downstream primers specific for each exon tagged with an unrelated chimeric 20-nucleotide sequence at the 5′ end of all primers (Shuber et al, 1995) (Table 2). Second round PCR was performed in a nested or hemi-nested condition using primers given in Table 2. Five microliters of I-PEP-PCR product was aliquoted to 25 μl of the specific PCR first-round mix: 0.2 mmol/L each dNTP; 1.5 mmol/L MgCl2; 0.4 μmol/L primers exons 5, 7, and 9; 0.6 μmol/L primers exons 6 and 8; 0.5 U of Taq Expand High Fidelity Polymerase; and 0.5 U of Anti-Taq Antibody (Gibco, Eggenstein, Germany). Each exon was amplified separately in a second-round PCR. Three microliters of first-round PCR product was aliquoted to 22 μl of mix containing 1.5 mmol/L MgCl2; 0.2 mmol/L each dNTP; 0.4 mmol/L primers exons 5 and 7; 0.6 mmol/L primers exons 6, 8, and 9; and 0.3 U of Taq Expand High Fidelity Polymerase. Cycling conditions were identical for first-round and second-round PCR: Step 1: 95° C for 2 minutes; Step 2: 95° C for 30 seconds; Step 3: 55° C for 45 seconds; Step 4: 72° C for 1 minute; Step 5: go to step 2, 29 times; Step 6: 72° C for 10 minutes; and Step 7: 4° C. Negative control reactions with water instead of DNA were included in each experiment. The presence and relative quantity of PCR product was ascertained by resolution on a 2% agarose gel. The primers of the nested PCR reaction were used for DNA sequencing.
DNA Sequencing
PCR products were purified using Microcon-100 Microconcentrators (Amicon Inc., Beverly, Massachusetts) and sequenced using the Big Dye Terminator Sequencing Kit (Applied Biosystems, Foster City, California) according to the manufacturers instructions. Sequence changes were confirmed by reamplifying from the original I-PEP-PCR and sequencing in the opposite direction. Repeated amplifications of the exons from another PEP aliquot of the same cell were performed to show that the mutation could be reproducibly identified in independent aliquots of the PEP. Sample chromatograms were analyzed in a blinded manner by two individuals. Seq Ed (PE Biosystems Foster City, California) and Sequencher software (Gene Codes, Inc., Ann Arbor, Michigan) were used to generate two or multiple sequence alignments, respectively. Differences between sequences were compared with the published wild-type sequence using the IARC TP53 mutation database (http://www.iarc.fr/p53/).
References
Arnold MM, Srivastava S, Fredenburgh J, Stockard CR, Myers RB, and Grizzle WE (1996). Effects of fixation and tissue processing on immunohistochemical demonstration of specific antigens. Biotech Histochem 71: 224–230.
Barnes DM, Hanby AM, Gillett CE, Mohammed S, Hodgson S, Bobrow LG, Leigh IM, Purkis T, MacGeoch C, and Spurr NK (1992). Abnormal expression of wild type p53 protein in normal cells of a cancer family patient. Lancet 340: 259–263.
Bassarova AV and Popov AA (1998). Immunohistochemical detection of p53: Effect of fixation and methods of antigen retrieval. Folia Histochem Cytobiol 36: 127–132.
Becker I, Becker KF, Rohrl MH, Minkus G, Schutze K, and Hofler H (1996). Single-cell mutation analysis of tumors from stained histologic slides. Lab Invest 75: 801–807.
Dietmaier W, Hartmann A, Wallinger S, Heinmoller E, Kerner T, Endl E, Jauch KW, Hofstadter F, and Ruschoff J (1999). Multiple mutation analyses in single tumor cells with improved whole genome amplification. Am J Pathol 154: 83–95.
Duddy SK, Gorospe S, and Bleavins MR (1998). Genetic analysis of multiple loci in microsamples of fixed paraffin-embedded tissue. Toxicol Sci 46: 317–323.
El-Hashemite N and Delhanty JD (1997). A technique for eliminating allele specific amplification failure during DNA amplification of heterozygous cells for preimplantation diagnosis. Mol Hum Reprod 3: 975–978.
Emmert-Buck MR, Bonner RF, Smith PD, Chuaqui RF, Zhuang Z, Goldstein SR, Weiss RA, and Liotta LA (1996). Laser capture microdissection [see comments]. Science 274: 998–1001.
Findlay I, Matthews P, and Quirke P (1998). Multiple genetic diagnoses from single cells using multiplex PCR: Reliability and allele dropout. Prenat Diagn 18: 1413–1421.
Findlay I, Ray P, Quirke P, Rutherford A, and Lilford R (1995). Allelic drop-out and preferential amplification in single cells and human blastomeres: Implications for preimplantation diagnosis of sex and cystic fibrosis. Hum Reprod 10: 1609–1618.
Forrester K, Lupold SE, Ott VL, Chay CH, Band V, Wang XW, and Harris CC (1995). Effects of p53 mutants on wild-type p53-mediated transactivation are cell type dependent. Oncogene 10: 2103–2111.
Garvin AM, Holzgreve W, and Hahn S (1998). Highly accurate analysis of heterozygous loci by single cell PCR. Nucleic Acids Res 26: 3468–3472.
Greenblatt MS, Bennett WP, Hollstein M, and Harris CC (1994). Mutations in the p53 tumor suppressor gene: Clues to cancer etiology and molecular pathogenesis. Cancer Res 54: 4855–4878.
Hachiya M, Chumakov A, Miller CW, Akashi M, Said J, and Koeffler HP (1994). Mutant p53 proteins behave in a dominant, negative fashion in vivo. Anticancer Res 14: 1853–1859.
Hahn S, Garvin AM, Di Naro E, and Holzgreve W (1998). Allele drop-out can occur in alleles differing by a single nucleotide and is not alleviated by preamplification or minor template increments. Genet Test 2: 351–355.
Hahn S, Zhong XY, Troeger C, Burgemeister R, Gloning K, and Holzgreve W (2000). Current applications of single-cell PCR. Cell Mol Life Sci 57: 96–105.
Huusko P, Castren K, Launonen V, Soini Y, Paakkonen K, Leisti J, Vahakangas K, and Winqvist R (1999). Germ-line TP53 mutations in Finnish cancer families exhibiting features of the Li-Fraumeni syndrome and negative for BRCA1 and BRCA2. Cancer Genet Cytogenet 112: 9–14.
Karlsen F, Kalantari M, Chitemerere M, Johansson B, and Hagmar B (1994). Modifications of human and viral deoxyribonucleic acid by formaldehyde fixation. Lab Invest 71: 604–611.
May P and May E (1999). Twenty years of p53 research: Structural and functional aspects of the p53 protein [published erratum appears in Oncogene 2000 Mar 23;19(13):1734]. Oncogene 18: 7621–7636.
Milner J and Medcalf EA (1991). Cotranslation of activated mutant p53 with wild type drives the wild-type p53 protein into the mutant conformation. Cell 65: 765–774.
Persson AE, Ling G, Williams C, Backvall H, Ponten J, Ponten F, and Lundeberg J (2000). Analysis of p53 mutations in single cells obtained from histological tissue sections. Anal Biochem 287: 25–31.
Ponten F, Williams C, Ling G, Ahmadian A, Nister M, Lundeberg J, Ponten J, and Uhlen M (1997). Genomic analysis of single cells from human basal cell cancer using laser-assisted capture microscopy. Mutat Res 382: 45–55.
Ray PF and Handyside AH (1996). Increasing the denaturation temperature during the first cycles of amplification reduces allele dropout from single cells for preimplantation genetic diagnosis. Mol Hum Reprod 2: 213–218.
Rechitsky S, Freidine M, Verlinsky Y, and Strom CM (1996). Allele dropout in sequential PCR and FISH analysis of single cells (cell recycling). J Assist Reprod Genet 13: 115–124.
Roehrl MH, Becker KF, Becker I, and Hofler H (1997). Efficiency of single-cell polymerase chain reaction from stained histologic slides and integrity of DNA in archival tissue. Diagn Mol Pathol 6: 292–297.
Schutze K and Lahr G (1998). Identification of expressed genes by laser-mediated manipulation of single cells. Nat Biotechnol 16: 737–742.
Shuber AP, Grondin VJ, and Klinger KW (1995). A simplified procedure for developing multiplex PCRs. Genome Res 5: 488–493.
Snabes MC, Chong SS, Subramanian SB, Kristjansson K, DiSepio D, and Hughes MR (1994). Preimplantation single-cell analysis of multiple genetic loci by whole-genome amplification. Proc Natl Acad Sci USA 91: 6181–6185.
Suarez-Quian CA, Goldstein SR, Pohida T, Smith PD, Peterson JI, Wellner E, Ghany M, and Bonner RF (1999). Laser capture microdissection of single cells from complex tissues. BioTechniques 26: 328–335.
Varley JM, McGown G, Thorncroft M, James LA, Margison GP, Forster G, Evans DG, Harris M, Kelsey AM, and Birch JM (1999). Are there low-penetrance TP53 alleles? Evidence from childhood adrenocortical tumors. Am J Hum Genet 65: 995–1006.
Villadsen R, Nielsen KV, Bolund L, and Briand P (2000). Complete loss of wild-type TP53 in a nontransformed human epithelial cell line is preceded by a phase during which a heterozygous TP53 mutant effectively outgrows the homozygous wild-type cells. Cancer Genet Cytogenet 116: 28–34.
Wallace-Brodeur RR and Lowe SW (1999). Clinical implications of p53 mutations. Cell Mol Life Sci 55: 64–75.
Walsh PS, Erlich HA, and Higuchi R (1992). Preferential PCR amplification of alleles: Mechanisms and solutions. PCR Methods Appl 1: 241–250.
Wells D, Sherlock JK, Handyside AH, and Delhanty JD (1999). Detailed chromosomal and molecular genetic analysis of single cells by whole genome amplification and comparative genomic hybridisation. Nucleic Acids Res 27: 1214–1218.
Williams C, Ponten F, Moberg C, Soderkvist P, Uhlen M, Ponten J, Sitbon G, and Lundeberg J (1999). A high frequency of sequence alterations is due to formalin fixation of archival specimens. Am J Pathol 155: 1467–1471.
Yagi N, Satonaka K, Horio M, Shimogaki H, Tokuda Y, and Maeda S (1996). The role of DNase and EDTA on DNA degradation in formaldehyde fixed tissues. Biotech Histochem 71: 123–129.
Zhang L, Cui X, Schmitt K, Hubert R, Navidi W, and Arnheim N (1992). Whole genome amplification from a single cell: Implications for genetic analysis. Proc Natl Acad Sci USA 89: 5847–5851.
Acknowledgements
Supported in part by the Deutsche Krebshilfe (to EH). Current address for Dr. Heinmöller: Institute of Pathology, Klinikum Kassel, Mönchebergstrasse 41-42, 34125 Kassel, Germany.
We thank Dr. Wolfgang Dietmaier for helpful discussions, Mrs. Wenyan Li, Mrs. Martha Magallanes, Mrs. Xuemin Li and Mrs. Sofia Loera for expert technical assistance, and Dr. S. Bates for the cells.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Heinmöller, E., Liu, Q., Sun, Y. et al. Toward Efficient Analysis of Mutations in Single Cells from Ethanol-Fixed, Paraffin-Embedded, and Immunohistochemically Stained Tissues. Lab Invest 82, 443–453 (2002). https://doi.org/10.1038/labinvest.3780437
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.3780437
