
Abstract
Endothelin-1 (ET-1) is a powerful mitogenic and/or anti-apoptotic peptide produced by many cancer cells. To evaluate the potential role of the endothelin system in glioblastoma we first determined the cellular distribution of the mRNA and proteins of the components of the endothelin system, preproendothelin-1 (PPET-1), endothelin-converting enzyme-1 (ECE-1), and ETA and ETB receptors in human glioblastoma tissue and glioblastoma cell lines. PPET-1, ECE-1, and ETA receptor were highly expressed in glioblastoma vessels and in some scattered glioblastoma areas whereas ETB receptor was mainly found in cancer cells. This suggests that glioblastoma vessels constitute an important source of ET-1 that acts on cancer cells via the ETB receptor. Four human glioblastoma cell lines expressed mRNA for all of the components of the ET-1 pathway. Bosentan, a mixed ETA and ETB receptor antagonist, induced apoptosis in these cell lines in a dose-dependent manner. Apoptosis was potentiated by Fas Ligand (APO-1L, CD95L), a pro-apoptotic peptide, only in LNZ308 cells, corresponding to the known functional Fas expression in these cell lines. LNZ308 cells also expressed the long and short forms of the cellular FLICE/caspase-8 inhibitory protein (FLIP). Bosentan and a protein kinase C inhibitor down-regulated short FLIP in these cells. ET-1 induced transient phosphorylation of extracellular signal-regulated kinase but did not induce long-term thymidine incorporation in LNZ308 glioblastoma cells. These results suggest that, in glioblastoma cells, ET-1, mainly acting via the ETB receptor, is a survival/anti-apoptotic factor produced by tumor vasculature, but not a proliferation factor, involving protein kinase C and extracellular signal-regulated kinase pathways, and stabilization of the short form of FLIP.
Similar content being viewed by others


Introduction
Endothelins (ET) (Yanagisawa et al, 1988) 1, 2, and 3 are a family of 21 amino-acid peptides enzymatically released from 200-residue prepolypeptides (Inoue et al, 1989) by furin-like activity to produce first inactive big ET (Denault et al, 1995). They are then cleaved by endothelin-converting enzyme (ECE-1) to yield the active peptides (Shimada et al, 1995). At least 4 isoforms, ECE-1a-d have been characterized (Valdenaire et al, 1999). ET act on two distinct high-affinity receptor subtypes, ETA (Arai et al, 1990) and ETB (Sakurai et al, 1990), which belong to the seven transmembrane G-protein–coupled receptor family. At physiologic concentrations, ET-1 and ET-2, but not ET-3, bind to ETA receptors, whereas all three ET ligands bind ETB receptors with a similar affinity. In addition to its vasoconstrictor activity, ET-1 is a potent autocrine/paracrine mitogen in many cell types, including neoplastic cells and astrocytes, involving the extracellular signal-regulated kinase (ERK) intracellular signaling pathway (Kusuhara et al, 1990, Shichiri et al, 1991; Teixeira et al, 2000). The expression of endothelins and endothelin receptors at the protein level has been shown in human glioma (Harland et al, 1995, 1998; Stiles et al, 1997; Tsutsumi et al, 1994).
Recently it has been suggested that ET-1, in addition to its mitogenic effects, may also regulate apoptosis in rat endothelial cells (Shichiri et al, 1997) and human smooth muscle cells (Wu-Wong et al, 1997). Apoptosis is an active cell death process that takes place in a wide spectrum of physiologic situations such as normal cell turnover, embryogenesis, and endocrine-dependent tissue atrophy. Interaction between the Fas receptor (CD95/APO-1), a member of the TNF-receptor superfamily, and the Fas ligand (FasL), triggers a pathway to cell death involving caspase activity. Although the expression of Fas and FasL has been demonstrated in glioblastoma (Saas et al, 1997), not all glioblastoma cell lines were able to respond to Fas engagement (Gratas et al, 1997). Tumor cell resistance to FasL-induced apoptosis has been described in different cancers by various mechanisms, including upregulation of caspase inhibitory molecules such as the pro–caspase-8 (FLICE)/caspase-8 inhibitory proteins (FLIP) (Irmler et al, 1997). We have recently shown that ET-receptor blockade by bosentan, a mixed ETA/ETB receptor antagonist, and two receptor subtype-specific antagonists, BQ123 (ETA selective) and BQ788 (ETB selective) sensitized Fas-expressing colon carcinoma cells, which were resistant to FasL-induced apoptosis, to FasL-mediated apoptosis (Peduto Eberl et al, 2000; 2000 in press).
These previous studies indicated a potential function for ET-1 in cell proliferation or as a cell survival factor. To evaluate the role of ET-1 in glioblastoma, we determined the expression of the complete ET-1 system in human glioblastoma and glioblastoma cell lines, then evaluated its role in human glioblastoma cell survival and proliferation.
Results
Endothelin System mRNA and Proteins in Human Glioblastoma
All of the components of the endothelin system were highly expressed in glioblastoma. In situ hybridization (ISH) localized preproendothelin-1 (PPET-1) and ECE-1 mRNA expression mainly to the tumor vasculature (Fig. 1a to c). ECE-1 mRNA signal colocalized with Ulex europaeus immunoreactivity (Fig. 1e) in endothelial cells and with ECE-1 immunoreactivity in tumor vasculature and in some tumor cells (Fig. 1f). PPET-1 mRNA was detected almost exclusively in endothelial cells (Fig. 1d), whereas ET-1 immunoreactivity was observed in α-smooth muscle actin- (α-SMA) immunoreactive cells in the vasculature, and in cancer cells (Fig. 1g), but not in all vessels (Fig. 1h). A perinuclear pattern of ET-1 immunoreactivity was evident in some glioblastoma areas (Fig. 1i).

Endothelin-1 (ET-1) and endothelin converting enzyme-1 (ECE-1) expression in human glioblastoma. In situ hybridization (ISH) was performed with the antisense probe for preproendothelin-1 (PPET-1) (a, b, d) and ECE-1 (c, e); and histochemistry with Ulex europaeus (e), ECE-1 (f), ET-1 (g, i), and α-smooth muscle actin (α-SMA) (h) antibodies. Darkfield (a, c) and bright-field illumination (b) of the same glioblastoma area in consecutive sections showed vascular expression of PPET-1 and ECE-1 mRNA. At higher magnification, PPET-1 (d) and ECE-1 (e) mRNA and ECE-1 immunoreactivity (f) were mainly expressed in endothelial cells as shown by colocalization of ECE-1 with Ulex europaeus (e). ET-1 immunoreactivity was found in α-SMA–immunoreactive cells, but not in glomeruloid structures (h) and in a perinuclear location in some glioblastoma cells (i). Scale bar = 100 μm in a to c, 20 μm elsewhere. Sense probes yielded no specific labeling and immunohistochemistry without primary antibody did not result in nonspecific reactivity (not shown).
ET-1 receptor distribution was evaluated using ISH (Fig. 2a to h) and [125I]–ET-1 binding autoradiography (Fig. 2i to m). ETA receptor mRNA was expressed in the tumor vasculature (Fig. 2a to d) and colocalized with CD31-immunoreactive endothelial cells (Fig. 2b), in microvessels (Fig. 2d), and in the glomeruloid α–SMA-immunoreactive vascular structures observed in glioblastoma and in few scattered tumor cells (Fig. 2c). ETB receptor mRNA was almost exclusively observed in glioblastoma cells (Fig. 2e to g), and restricted to a few scattered endothelial cells (Fig. 2h). To assess the presence of functional ETA and ETB receptors in human glioblastoma, we performed [125I]–ET-1 binding in frozen samples (Fig. 2i to m). [125I]–ET-1 (Fig. 2i) bound specifically to glioblastoma cells and vessels. Nonspecific binding that remained after displacement with ET-1 was uniformly low (Fig. 2j). Identification of receptor subtype was performed in consecutive sections by competition with the selective ETB receptor agonist sarafotoxin 6c, which showed the presence of the ETA receptor (Fig. 2k), and also with the selective ETA receptor antagonist, BQ123, which indicated the presence of the ETB receptor (Fig. 2l). The presence of ETA receptor was observed in tumor vasculature and the ETB receptor was found in glioblastoma cells, confirming the ISH results. This pattern of distribution was very similar in all tumor specimens. Table 1 summarizes the distribution of the endothelin system in human glioblastoma.

ETA and ETB receptors in human glioblastoma. Darkfield (a, d) and bright-field illumination (c, e, f) of ISH performed with the anti-sense probes for ETA receptors (a to d) and ETB receptors (e to h) in human glioblastoma. Autoradiographic [125I]–ET-1 (100 pm) binding was performed in frozen consecutive sections (i to m). Whereas the ETA receptor showed a vascular distribution pattern demonstrated by colocalization with CD31 (b) immunoreactivity, the ETB receptor (e) displayed a homogeneous distribution pattern in cancer cells. At high magnification, ETA receptor mRNA was expressed in the tumor vasculature (c, d) and ETB receptor in tumor cells (g), and some endothelial cells (h). ET-1 binding was used to ascertain functional receptor expression. i, Total ET-1 binding; j, nonspecific ET-1 binding; k, ETA receptor binding in the presence of the ETB receptor antagonist sarafotoxin 6c; l, ETB receptor binding in the presence of the ETA receptor antagonist BQ123; m, bright-field illumination of the same glioblastoma area. Scale bar = 100 μm in a, b, e, f, i to m; 20 μm in c, d, g, h. Sense probes yielded no specific labeling and immunohistochemistry without primary antibody did not result in nonspecific reactivity (not shown).
Role of ET-1 in Human Glioblastoma
RT-PCR analysis of four human glioblastoma cell lines, LN18, LN215, LNZ308, and LN319, demonstrated the presence of all of the components of the endothelin system, including the ECE-1a-d isoforms, PPET-1, PPET-3, and ETA and ETB receptors (Fig. 3). These cells secreted low levels of ET-1 in the presence of FCS over 48 hours (LN18: 8.9 pg/ml, LN215: 1.7 pg/ml, LNZ308: 2.8 pg/ml, and LN319: <0.3 pg/ml). ET-1 secretion was below detection limits in the absence of FCS. These cells expressed low levels of ECE-1 as assessed by immunohistochemistry and western blotting (data not shown).

Components of the endothelin system in human glioblastoma cell lines. RNA from four glioblastoma cell lines, LN18, LN215, LNZ308, and LN319, were used for RT-PCR analysis for PPET-1, PPET-3, ECE-1, ETA and ETB receptors, and GADPH, using the specific primers described in Table 2. Twenty-eight cycles of amplification gave each transcript at the expected size. Lane 1: 354 bp (PPET-1); lane 2: 445 bp (PPET-3), lane 3: 576 bp (ETA receptor); lane 4: 477 bp (ETB receptor); lane 5: 459 bp (ECE-1); lane 6: 353 bp (ECE-1a); lane 7: 347 bp (ECE-1b); lane 8: 348 bp (ECE-1c); lane 9: 369 bp (ECE-1d); and lane 10: 403 bp (GADPH). Molecular weight markers are Φ174DNA/HaeIII. The high molecular weight band in the ETB receptor lane is genomic DNA amplification.
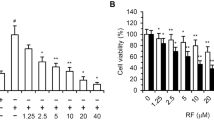
Increasing concentrations of bosentan, a mixed ETA/ETB receptor antagonist, induced apoptosis in the four glioblastoma cell lines in a dose-dependent manner (Fig. 4, open symbols). Addition of FasL-containing supernatants increased apoptosis only in LNZ308 cells. LN18 cells were sensitive to FasL-containing supernatants in the absence of bosentan, whereas addition of FasL-containing supernatants to LN215 and LN319 glioblastoma cells did not potentiate bosentan-induced apoptosis. (Fig. 4, closed symbols). The addition of Neuro2A-control medium alone or in the presence of bosentan had no effect in any conditions (not shown). Rat glioblastoma Fas-positive F98 and Fas-negative F98ZH (Ambar et al, 1999) cells exposed to FasL-containing supernatants in the presence of bosentan were used as controls for the Fas/FasL dependency of the system (not shown).

Effect of bosentan and FasL on glioblastoma cell apoptosis. Fifty percent-confluent glioblastoma cells were incubated with increasing concentrations of bosentan (10 to 450 μm) in the absence (□) or in the presence (▪) of FasL-containing supernatant from Neuro2A cells. Apoptosis was evaluated after 24 hours of incubation. One representative experiment out of three is shown.
Fas/FasL-responsive bosentan-sensitive LNZ308 cells were used to evaluate the roles of ET-1 and bosentan in glioblastoma apoptosis. Addition of 40 nm ET-1 (not shown), 0.4 nm ET-1 (Fig. 5a) or 4 pm ET-1 (not shown) induced rapid and transient p44/42 mitogen-associated protein (MAP) kinase/ERK phosphorylation in LNZ308 cells, demonstrating the presence of functional receptors and intracellular pathways in these cells. Addition of increasing concentrations of ET-1 for 3 hours, 5 hours, or 48 hours to glioblastoma cells, after 24 hours of FCS deprivation, did not increase thymidine incorporation (Fig. 5b), demonstrating that, at least in these glioblastoma cell lines, ET-1 is not importantly involved in sustained glioblastoma cell proliferation. Identical information was obtained using LN18, LN215, and LN319 cells (not shown).

Response of glioblastoma cells to exogenous ET-1. a: Extracellular signal-regulated kinase (ERK) phosphorylation. After 24 hours of FCS deprivation, LNZ308 cells were incubated with 0.4 nm ET-1. Lane 1: no ET-1; lane 2: FCS, 120 minutes; lane 3: ET-1, 5 minutes; lane 4: ET-1, 10 minutes; lane 5: ET-1, 30 minutes; lane 6: ET-1, 60 minutes; lane 7: ET-1, 120 minutes. b: DNA synthesis. After 24 hours of FCS deprivation, LNZ308 cells were incubated simultaneously with increasing concentrations of ET-1 and [3H]-thymidine for 48 hours. Values are the means ± sem of two independent determinations performed in triplicate wells.
Activation of the Fas death pathway involves the activation of FLICE, which can be antagonized by cellular inhibitors of caspase activation. Cellular FLIP is an inhibitory molecule potentially involved in resistance to FasL-induced apoptosis. Two forms of FLIP, a long and a short form, have been described (Irmler et al, 1997). To evaluate the possibility that bosentan sensitization to FasL-mediated apoptosis in LNZ308 cells may involve FLIP down-regulation or inactivation, we analyzed the expression of FLIP in glioblastoma cells. Glioblastoma cell lines expressed the short and long forms of cellular FLIP, as assessed by RT-PCR (Fig. 6) and expressed caspase-8, as assessed by western blotting (not shown). The short and long forms of FLIP mRNA were still expressed (not shown) in bosentan-treated cells 24 hours after exposure. However, in LNZ308 cells exposed to high concentrations of bosentan alone or in combination with FasL-containing supernatant, the concentration of the short form, but not the long form, of the FLIP protein was decreased (Fig. 7a). The highest concentration of bosentan reproduced the effect of 10 μm of the protein kinase C (PKC) inhibitor, bisindolylmaleimide IX (Fig. 7b).

Expression of long and short forms of cellular pro–caspase-8 (FLICE)/caspase-8 inhibitory protein (FLIP) by RT-PCR in four human glioblastoma cell lines. RT-PCR analysis for short and long FLIP was performed on total RNA extracted from the four glioblastoma cell lines using the specific primers described in Table 2. M: DNA marker. Two independent experiments gave similar results. Lane 1: LN18 cells; lane 2: LN215 cells; lane 3: LNZ308 cells; lane 4: LN319 cells. Molecular weight markers are Φ174DNA/HaeIII.

Expression and regulation of caspase-8 and of long and short forms of cellular FLIP in the human glioblastoma cell line LNZ308. Expression of caspase-8 and of the long and short forms of FLIP by bosentan and FasL (a) or the protein kinase C (PKC) inhibitor bisindolylmaleimide IX (b) were analyzed by Western blotting. a, Short and long FLIP in LN308 cells exposed to bosentan and FasL for 24 hours. b, Short FLIP in LN308 cells exposed for 24 hours to the PKC inhibitor bisindolylmaleimide IX. Two independent experiments gave similar results.
Discussion
ET-1 was previously shown to be produced by human cancer cell lines and may be a growth promoting and survival factor for tumor and stromal cells. The expression of ET-1, ET-3, and their receptors has been demonstrated in the brain, and in human glioblastoma (Harland et al, 1995, 1998; Tsutsumi et al, 1994). Glioblastoma also express the death-inducing receptor Fas (Saas et al, 1997). However, not all human glioblastoma cell lines are sensitive to Fas-induced death while expressing Fas (Gratas et al, 1997). Recently, we have shown that colon cancer overexpresses all of the components of the ET-1 system (Egidy et al, in press) and that ET-1 is involved in the resistance of colon tumor cell lines to Fas-induced death (Peduto Eberl et al, 2000; 2000 in press). We hypothesized that ET-1 may also be involved in the proliferation or survival of glioblastoma, either through an autocrine effect on tumor cells or a paracrine effect on cancer-associated stromal cells. To evaluate this hypothesis, we examined the expression and potential function of the endothelin system components, including PPET-1, ECE-1, and ETA and ETB receptors in human glioblastoma and human glioblastoma cell lines.
To our knowledge, no studies have been reported on the precise cellular distribution of ECE-1 in relation to the other components of the ET-1 pathway, PPET-1, ETA and ETB receptors, in human glioblastoma. Our results demonstrated that mRNA for the substrate, PPET-1, its converting enzyme, ECE-1, and ETA receptors were mainly expressed in tumor vessels, whereas ETB receptors were almost exclusively expressed by tumor cells. The expression of ECE-1 protein was mainly vascular, although it could be detected in some locations in the tumors. A higher proportion of cells was detected with the ET-1 antibody than with the PPET-1 probes. This suggests immunoreactivity of receptor-bound ET-1, either at the cell membrane or to internalized receptors, and a paracrine role for vascular-derived ET-1. The distribution of ETA and ETB receptors determined by ET-1 binding corresponded to the distribution observed using ISH. ETA receptors were highly expressed in vascular structures, α–SMA-immunoreactive cells, and endothelial cells, which confirms previously reported results (Harland et al, 1995, 1998; Tsutsumi et al, 1994), and in the glomeruloid structures of glioblastoma vasculature. ETB receptors were found mainly in glioblastoma cells, and in only a very few endothelial cells. Thus, major differences are observed in cellular distribution, and consequently, the functions, of the ET-1 system between glioblastoma and colon carcinoma, in which ETA and ETB receptors were mainly observed in tumor-associated vasculature, myofibroblasts, and endothelial cells, and PPET-1 and ECE-1 were expressed in cancer cells (Egidy et al, in press).
What is the role of the endothelin system in glioblastoma? We used human glioblastoma cell lines to address this question,. All components of the ET-1 system were detected by RT-PCR in four human glioblastoma cell lines. These cells secreted low levels of ET-1, corresponding to the low expression of PPET-1 observed by ISH in glioblastoma. They responded to ET-1 by transiently phosphorylating ERK, demonstrating the presence of functional ET-1 receptors and intracellular signaling pathways in the glioblastoma cell lines. ERK phosphorylation in response to ET-1 has been previously demonstrated in primary rat astrocytes (Sasaki et al, 1998; Teixeira et al, 2000), thus, astrocyte-derived human tumor cells have maintained this response to ET-1. However, ET-1 is not an important growth factor for glioblastoma cells for long-term proliferation. The ability of ET-1 to stimulate ERK phosphorylation was mediated by PKC activation in airway smooth muscle cells (Whelchel et al, 1997), but not in Rat-1 cells (Cadwallader et al, 1997).
Bosentan (Clozel et al, 1993), a mixed ETA/ETB receptor antagonist, induced apoptosis in all glioblastoma cell lines in a dose-dependent manner, but potentiated FasL-mediated apoptosis only in the LNZ308 cells, at concentrations below those needed to induce apoptosis with bosentan alone. In LN18 cells, FasL induced death in the absence of bosentan, whereas in LN215 and LN319 cells, FasL had no effect. It was previously shown that the cell lines used in this study express Fas and FasL, and that Fas engagement induced apoptosis only in LN18 cells (Gratas et al, 1997). Our results confirm those findings. In rat glioblastoma F98 cells, which express functional Fas, bosentan potentiated the FasL effect, whereas in Fas-negative F98ZH cells (Ambar et al, 1999), no potentiation of the effect was observed, demonstrating the Fas/FasL dependency of bosentan effects in glioblastoma.
The death receptor Fas is a member of the tumor necrosis factor family. Upon interaction with its ligand, it activates caspase-8 and induces cell death. However, despite expression of Fas and caspase, death signals are frequently interrupted by anti-apoptotic modulators, including caspase inhibitory proteins such as FLIP (Irmler et al, 1997). Glioblastoma cell lines expressed caspase-8 protein and the long and short forms of FLIP, assessed by RT-PCR. Treatment of LNZ308 cells with bosentan alone or in combination with FasL resulted in a decreased level of only the short form of FLIP protein, whereas short FLIP mRNA was not suppressed in these cells. This suggests that either translation is diminished or short FLIP degradation is increased. A similar effect on short FLIP was observed after treatment with a PKC inhibitor, suggesting that the bosentan effect is mediated by inhibition of PKC function. However, the high concentrations of bosentan necessary to obtain this effect indicate that the bosentan action is not likely to be mediated by cell membrane ET-1 receptors, but may involve intracellular components. In rat (Peduto Eberl et al, 2000) and human (Peduto Eberl et al, 2000 in press) colon cancer cell lines, blockade of caspase activity by zVAD-fmk had no effect on cells treated with bosentan in the absence of FasL, whereas apoptosis was completely inhibited by zVAD-fmk in the presence of both bosentan and FasL, involving PKC blockade but not the ceramide pathway. In human colon carcinoma cells, bosentan treatment did not modify either short or long forms of FLIP (Peduto Eberl et al, 2000 in press).
In conclusion, the different components of the endothelin system were examined in human glioblastoma. Expression of ECE-1 and PPET-1 in tumor vasculature provides a local source of ET-1 involved in a paracrine role in tumor cells via ETB receptors and an autocrine function in tumor-associated vasculature via ETA receptors. In human glioblastoma cell lines, the ET-1 system was not involved in tumor proliferation but ET-1 was a survival factor, possibly acting through ERK phosphorylation and the PKC pathway, resulting in a stabilization of the short form of FLIP protein.
Materials and Methods
Human Tissues
Human glioblastoma tissue was randomly selected form surgical diagnostic biopsies and subtotal resection specimens, and either fixed in 4% buffered paraformaldehyde and embedded in paraffin, or frozen in liquid nitrogen and stored at −80° C. Paraffin-embedded (n = 8) and unfixed frozen (n = 4) samples were used for ISH and immunohistochemistry and for ET-1 binding, respectively.
Immunohistochemistry
Paraffin-embedded sections (5 μm thick) of human glioblastoma were deparaffinized in xylene and isopropanol and endogenous peroxidase was inactivated in 3% hydrogen peroxide in methanol. Sections were washed in PBS, incubated with primary antibodies: human anti-CD31 and anti-CD68 (Dako, Zug, Switzerland), anti–α-SMA (Sigma, Buchs, Switzerland), monoclonal anti–ET-1 (ABR; Affinity Bioreagents, Alexis Corporation, Läufelingen, Switzerland), polyclonal anti–ET-1 (Peninsula, Brunschwig, Basel, Switzerland), anti–ECE-1 (antiserum 473-17-A; (Korth et al, 1999), or to Ulex Europaeus (Vector, Alexis Corporation) and subsequently exposed either to avidin-biotin complex (ABC; Dako) or to peroxidase-conjugated secondary immunoglobulins, according to the manufacturers' instructions. Peroxidase activity was visualized using 0.035% diaminobenzidine (Fluka, Buchs, Switzerland) as a chromogen and slides were counterstained with hematoxylin. Reactions performed without the primary antibody were used as controls for nonspecific reactions (not shown).
ISH and [125I]–ET-1 Binding
ISH for the paraffin sections was performed as previously described (Egidy et al, 2000), with antisense and sense probes for PPET-1, ECE-1, and ETA and ETB receptors. Sense probes yielded no specific labeling in any tissue (not shown). Ulex Europaeus and CD31 labeling were performed on some sections immediately after ISH, before immersion into the Kodak NTB2 photographic emulsion. Slides were exposed for 4 weeks (ECE-1 and ETA and ETB receptor probes) or 8 weeks (PPET-1 probe). Frozen glioblastoma sections (7 μm thick) were used to evaluate binding of [125I]–ET-1 as previously described (Egidy et al, 2000). Briefly, consecutive sections fixed in 4% formaldehyde in PBS were exposed to 100 pm [125I]–ET-1 (2125 Ci/mmol) in 50 mm Tris-HCl buffer, pH 7.5, containing 120 mm NaCl, 5 mm MgCl2, 40 mg/l of bacitracin, 1 mm phosphoramidon, and 1% BSA (fraction V, protease-free). Nonspecific binding was determined similarly in consecutive sections incubated in the presence of 1 μm unlabeled ET-1 (Bachem, Bubendorf, Switzerland). To characterize the receptor subtypes, sections were incubated in the presence of 1 μm BQ123, a selective ETA receptor antagonist, or 0.2 μm sarafotoxin 6c, a selective ETB receptor agonist.
Cell Culture and Treatments
The human glioblastoma cell lines LN18, LN215, LNZ308, and LN319 were cultured in DMEM with 4.5 g/l of glucose, supplemented with 10% FCS. Cells were grown as monolayers to 50% confluence, washed, and incubated at 37° C with the endothelin-receptor antagonist, bosentan (kindly supplied by Dr. M. Clozel, Actelion, Basel, Switzerland), in the presence or absence of Neuro2A FasL-containing supernatant (Rensing-Ehl et al, 1995) (2 volumes of culture medium:10% FCS and 1 volume of FasL-containing supernatant) for 24 hours or 48 hours. Rat glioblastoma Fas-positive F98 and Fas-negative F98ZH cells (Ambar et al, 1999) were used as controls. Cell culture reagents were purchased from Gibco-BRL (Life Technologies, Basel, Switzerland) and bisindolylmaleimide IX from Calbiochem (Juro, Luzern, Switzerland).
Thymidine Incorporation
Cells were grown to 75% confluence. FCS was then removed from the culture for 24 hours. Increasing concentrations of ET-1 (Bachem) were added together with 0.2 μCi/well of [3H]-thymidine (Amersham Pharmacia, Dübendorf, Switzerland) for 3, 5, or 48 hours, and incorporation was quantitated as previously described (Juillerat-Jeanneret et al, 1992).
Determination of p44/42 MAP Kinase/ERK Phosphorylation
Cells were grown in DMEM containing 10% FCS, deprived of serum for 24 hours, and exposed either to ET-1 (Bachem) for 5 to 120 minutes or to 10% FCS for 120 minutes. Cell cultures were extracted using 0.1% Triton X-100 in the presence of protease inhibitors (Roche Boehringer, Rotkreuz, Switzerland) and the extracts were submitted to electrophoresis. After transfer, the membrane was probed using a phospho-p44/42 MAP kinase monoclonal antibody (clone E10; New England BioLabs, Bioconcepts, Allschwil, Switzerland).
Cell Death Evaluation
Apoptosis was detected using the Cell Death Detection ELISAPLUS (Roche Boehringer), a photometric enzyme-linked immunoassay for quantitative in vitro determination of cytoplasmic histone-associated DNA-fragments, as previously described (Peduto Eberl et al, 2000).
Measurement of Endothelin Secretion
The quantitative determination of ET-1 in cell culture supernatants at 24 hours or 48 hours was performed using a chemiluminescent enzyme-linked immunoassay (QuantiGlo; R&D Systems, Wiesbaden, Germany), according to the supplier's instructions. Cross-reactivity with human big–ET-1, ET-2, and ET-3 was less than 0.1%, 27.4%, and 7.8%, respectively, according to the manufacturer's specifications.
mRNA Analysis by RT-PCR
Total RNA was isolated from cell cultures using the Trizol reagent (Gibco-BRL). RT-PCR was performed according to standard procedures. Sequences of the primers used for each cDNA are given in Table 2. As controls, amplification reactions were performed with pairs of primers specific for human glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Control reactions for RT-PCR analyses were carried out from non–reverse-transcribed RNA samples. No amplification was observed for any of the RNA samples tested (not shown). Twenty-eight cycles were carried out and amplified products were analyzed on 2% agarose gel.
Determination of Long and Short Forms of FLIP and Caspase-8 by Western Blotting
Confluent cell cultures were extracted using 0.1% Triton X-100 and protease inhibitors, and cell extracts were submitted to electrophoresis under nonreducing conditions on a 12% polyacrylamide gel. After transfer, the membrane was probed using the human FLIP AL148 polyclonal antibody, which was raised against a peptide spanning the amino acid sequences 2 to 26, which are common to both the short and long forms of FLIP (Irmler et al, 1997). Caspase-8 determination was performed as previously described (Irmler et al, 1997).
Statistical Analysis
Means and standard deviation (sd) were calculated. Statistical significance was assessed using a two-tailed Student's t test. Experiments were repeated at least three times with comparable information, unless otherwise indicated.
References
Ambar BB, Frei K, Malipiero U, Morelli AE, Castro MG, Lowenstein PR, and Fontana A (1999). Treatment of experimental glioma by administration of adenoviral vectors expressing Fas ligand. Human Gene Therapy 10: 1641–1648.
Arai H, Hori S, Aramori I, Ohkubo H, and Nakanishi S (1990). Cloning and expression of a cDNA encoding an endothelin receptor. Nature 348: 730–732.
Cadwallader K, Beltman J, McCormick F, and Cook S (1997). Differential regulation of extracellular signal-regulated protein kinase 1 and Jun N-terminal kinase 1 by Ca2+ and protein kinase C in endothelin-stimulated Rat-1 cells. Biochem J 321: 795–804.
Clozel M, Breu V, Burri K, Cassal JM, Fischli W, Gray GA, Hirth G, Loffler BM, Muller M, Neidhart W, and Ramuz H (1993). Pathophysiological role of endothelin revealed by the first orally active endothelin receptor antagonist. Nature 365: 759–761.
Denault JB, Claing A, D'Orléans-Juste P, Sawamura T, Kido T, Masaki T, and Leduc R (1995). Processing of proendothelin-1 by human furin convertase. FEBS Lett 362: 276–280.
Egidy G, Juillerat-Jeanneret L, Jeannin JF, Korth P, Bosman FT, and Pinet F (in press, 2000). Modulation of human colon tumor-stromal interactions by the endothelin system. Am J Pathol.
Egidy G, Juillerat-Jeanneret L, Korth P, Bosman FT, and Pinet F (2000). The endothelin system in human colon. Am J Physiol 279: G211–G222.
Gratas C, Tohma Y, van Meir EG, Klein M, Tenan M, Ishii N, Tachibana O, Kleihues P, and Ohgaki H (1997). Fas ligand expression in glioblastoma cell lines and primary astrocytic brain tumors. Brain Pathol 7: 863–869.
Harland SP, Kuc RE, Pickard JD, and Davenport AP (1995). Characterization of endothelin receptors in human brain cortex, gliomas and meningiomas. J Cardiovasc Pharm 26: S408–S411.
Harland SP, Kuc RE, Pickard JD, and Davenport AP (1998). Expression of endothelin A receptors in human gliomas and meningiomas, with high affinity for the selective antagonist PD156707. Neurosurgery 43: 890–898.
Inoue A, Yanagisawa M, Kimura S, Kasuya Y, Miyauchi T, Goto K, and Masaki T (1989). The human endothelin family: Three structurally and pharmacologically distinct isopeptides predicted by three separate genes. Proc Natl Acad Sci USA 86: 2863–2867.
Irmler M, Thome M, Hahne M, Schneider P, Hofman K, Steiner V, Bodmer JL, Schröter M, Burns K, Mattmann C, Rimoldi D, French LE, and Tschopp J (1997). Inhibition of death receptor signals by cellular FLIP. Nature 388: 190–195.
Juillerat-Jeanneret L, Aguzzi A, Wiestler OD, Darekar P, and Janzer RC (1992). Dexamethasone selectively regulates the activity of enzymatic markers of cerebral endothelial cell lines. In Vitro Cell Dev Biol 28A: 537–543.
Korth P, Bohle RM, Corvol P, and Pinet F (1999). Cellular distribution of endothelin converting enzyme-1 in human tissues. J Histochem Cytochem 47: 447–462.
Kusuhara M, Yamaguchi K, Nagasaki K, Hayashi C, Suzaki A, Hori S, Handa S, Nakamura Y, and Abe K (1990). Production of endothelin in human cancer cell lines. Cancer Res 50: 3257–3261.
Peduto Eberl L, Egidy G, Pinet F, and Juillerat-Jeanneret L (In press, 2000). Endothelin receptor blockade potentiates FasL-induced apoptosis in colon carcinoma cells via the protein kinase C pathway. J Cardiovasc Pharm.
Peduto Eberl L, Valdenaire O, SaintGiorgio V, Jeannin JF, and Juillerat-Jeanneret L (2000). Endothelin receptor blockade potentiates FasL-induced apoptosis in rat colon carcinoma cells. Int J Cancer 86: 182–187.
Rensing-Ehl A, Frei K, Flury R, Matiba B, Mariani SM, Weller M, Aebischer P, Krammer PH, and Fontana A (1995). Loco-regional Fas/APO-1 (CD95) ligand-mediated tumor cell killing in vivo. Eur J Immunol 25: 2253–2258.
Saas P, Walker PR, Hahne M, Quiquerez AL, Schnuriger V, Perrin G, French L, Van Meir EG, de Tribolet N, Tschopp J, and Dietrich PY (1997). Fas ligand expression by astrocytoma in vivo: Maintaining immune privilege in the brain? J Clin Invest 99: 1173–1178.
Sakurai T, Yanagisawa M, Takuwa Y, Miyazaki H, Kimura S, Goto K, and Masaki T (1990). Cloning of a cDNA encoding a non-isopeptide-selective subtype of the endothelin receptor. Nature 348: 732–735.
Sasaki Y, Hori S, Oda K, Okada T, and Takimoto M (1998). Both ETA and ETB receptors are involved in mitogen-activated protein kinase activation and DNA synthesis of astrocytes: Study using ETB receptor-deficient rats (aganglionosis rats). Eur J Neurosci 10: 2984–2993.
Shichiri M, Hirata Y, Nakajima T, Ando K, Imai T, Yanagisawa M, Masaki T, and Marumo F (1991). Endothelin-1 is an autocrine/paracrine growth factor for human cancer cell lines. J Clin Invest 87: 1867–1871.
Shichiri M, Kato H, Marumo F, and Hirata Y (1997). Endothelin-1 as an autocrine/paracrine survival factor for endothelial cells. Hypertension 30: 1198–1203.
Shimada K, Matsushita Y, Wakabayashi K, Takahashi M, Matsuba A, Iijima Y, and Tanzawa K (1995). Cloning and functional expression of human converting enzyme cDNA. Biochem Biophys Res Com 207: 807–812.
Stiles JD, Ostrow PT, Balos LL, Greenberg SJ, Plunkett R, Grand W, and Heffner RR (1997). Correlation of endothelin-1 and transforming growth factor beta-1 with malignancy and vascularity in human glioblastoma. J Neuropath Exp Neurol 56: 435–439.
Teixeira A, Chaverot N, Strosberg AD, and Cazaubon S (2000). Differential regulation of cyclin D1 and D3 expression in the control of astrocyte proliferation induced by endothelin-1. J Neurochem 74: 1034–1040.
Tsutsumi K, Niwa M, Kitagawa N, Yamaga S, Anda T, Himeno A, Sato T, Khalid H, Taniyama K, and Shibata S (1994). Enhanced expression of an endothelin ETA receptor in capillaries from human glioblastoma: A quantitative receptor autoradiographic analysis using a radioluminographic imaging plate system. J Neurochem 63: 2240–2247.
Valdenaire O, Lepailleur-Enouf D, Egidy G, Touhard A, Barret A, Vranckx R, and Tougard C (1999). A fourth isoform of endothelin converting enzyme (ECE-1) is generated from an additional promoter. Molecular cloning and characterization. Eur J Biochem 264: 341–349.
Whelchel A, Evans J, and Posada J (1997). Inhibition of ERK activation attenuates endothelin-stimulated airway smooth muscle cell proliferation. Am J Resp Cell Mol Biol 16: 589–596.
Wu-Wong J, Chiou WJ, Dickinson R, and Opgenorth TJ (1997). Endothelin attenuates apoptosis in human smooth muscle cells. Biochem J 328: 733–737.
Yanagisawa M, Kurihara M, Kimura H, Tomobe S, Kobayashi Y, Mitsui M, Yazaki Y, Goto K, and Masaki T (1988). A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature 332: 411–415.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work has been supported by the Swiss National Science Foundation (Grant 32.045908.95), the Swiss League and Research Against Cancer (Grant SKL 353-9-1996), the Swiss Society for Multiple Sclerosis, the Ministère des Affaires Etrangères, the Swiss Program “Cotutelle de thèse” and the French Embassy in Switzerland, the Foundation Cino and Simone del Duca, the Foundation pour la Recherche Médicale, and the Ministère Français de la Recherche et de l’Enseignement (ACC-SV9).
We would like to thank Ms. S. Gross, Ms. P. Fioroni, and Ms. R. Bovey for excellent technical assistance; Dr. M. Clozel, Actelion, Basel, Switzerland, for the gift of bosentan; and Drs. Jürg Tschopp, J. D. Aubert, and F. T. Bosman for very helpful discussion and suggestions.
Rights and permissions
About this article
Cite this article
Egidy, G., Eberl, L., Valdenaire, O. et al. The Endothelin System in Human Glioblastoma. Lab Invest 80, 1681–1689 (2000). https://doi.org/10.1038/labinvest.3780178
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.3780178
This article is cited by
-
Overexpression of endothelin B receptor in glioblastoma: a prognostic marker and therapeutic target?
BMC Cancer (2018)
-
Differential somatostatin, CXCR4 chemokine and endothelin A receptor expression in WHO grade I–IV astrocytic brain tumors
Journal of Cancer Research and Clinical Oncology (2018)
-
Renin angiotensin system and its role in biomarkers and treatment in gliomas
Journal of Neuro-Oncology (2018)
-
The brain tissue response to surgical injury and its possible contribution to glioma recurrence
Journal of Neuro-Oncology (2016)
-
Endothelin 1 in cancer: biological implications and therapeutic opportunities
Nature Reviews Cancer (2013)
