
Abstract
Molecular typing in Creutzfeldt–Jakob disease (CJD) relies on the detection of distinct protease-resistant prion protein (PrPSc) core fragments, which differ in molecular mass or glycoform ratio. However, the definition and correct identification of CJD cases with a co-occurrence of PrPSc types remains a challenge. With antibodies recognizing a linear epitope in the octapeptide repeat PrP region, supposed to distinguish between the two major PrPSc isoforms (ie, types 1 and 2), it was recently shown that all type 2 cases display an associated band with a type 1 migration pattern, which led to the conclusion that multiple PrPSc types regularly coexist in CJD. We studied brain samples from 53 sporadic CJD and 4 variant CJD subjects using a high-resolution electrophoresis, a wide range of proteinase K (PK) activities, the ‘type 1-selective’ antibody 12B2, and several unselective antibodies. We found that the type 1-like band detected by 12B2 in all CJD subtypes associated with PrPSc type 2 is not a PK-resistant PrPSc core but rather matches the physicochemical properties of partially cleaved fragments, which result from the several PK cleavage sites included in the N-terminal portion of PrPSc. Furthermore, using gels with high resolution and a relatively high PK activity, we were able to increase the detection sensitivity of either type 1 or 2, when coexisting, to amount corresponding to 3–5% of the total PrPSc signal (ie, weak band of one type/total PrPSc). Our results show that there are many pitfalls associated with the use of ‘type 1 selective’ antibodies for CJD typing studies and that co-occurrence of PrPSc types in CJD is not the rule. The present results further validate the rationale basis of current CJD classification and the qualitative nature of molecular typing in CJD.
Similar content being viewed by others

Main
Transmissible spongiform encephalopathies (TSEs), or prion diseases, are a phenotypically heterogeneous group of neurodegenerative disorders of infectious, sporadic, or genetic origin, affecting humans and animals. The pathogenesis of TSEs is linked to the cellular prion protein (PrPC), a host-encoded, copper and membrane-bound glycoprotein of unknown function.1 There is no agent replication or transmission of infectivity in the absence of PrPC expression.2 Moreover, an abnormal isoform (PrPSc) of PrPC specifically accumulates in the nervous system during disease progression and represents the hallmark of the disease.1 PrPC is thought to be converted into PrPSc through a post-translational event associated with an increase in β-sheet secondary structure and a greater aggregation of the protein.3, 4 As a consequence, PrPSc becomes insoluble in non-denaturing detergent and, in a significant proportion,5 also acquires a partial protease resistance, a distinctive property that is commonly used to distinguish PrPSc from PrPC. Incubation of a TSE-infected brain homogenate with proteinase K (PK) under conditions leading to a complete degradation of PrPC generates an N-terminal truncated form of PrPSc commonly referred to as PrP27-30.6
Creutzfeldt–Jakob disease (CJD), the most common human TSE, like scrapie, the prototype of animal TSEs, comprises a broad spectrum of clinicopathological variants.7, 8 The existence of multiple strains of the infectious agent in conjunction with host genetic factors such as PRNP mutations or polymorphisms are thought to be responsible for TSE phenotypic heterogeneity.9 In humans, disease susceptibility and phenotypic expression are influenced by PRNP mutations and by the polymorphism at codon 129 that encodes either methionine (M) or valine (V).10, 11, 12 Distinct agent strains have been demonstrated13, 14, 15, 16 but have yet to be characterized in full.
Although uncertainties remain on the molecular basis of TSE strains and the relationship between strains and PrP, several lines of evidence indicate that PrPSc exists in a variety of molecular subtypes showing distinctive physicochemical properties13, 17, 18, 19, 20, 21, 22, 23, 24, 25 (for reviews about more recent studies see Parchi et al26 and Baron et al27). Most significantly, PrPSc molecules derived from different strains or disease variants with distinct pathology often vary in their N-terminal site of PK cleavage. Based on differences in gel mobility and N-terminal sequence of the core fragments (ie, PrP27–30) generated by PK, Parchi et al20, 21 originally identified two major human PrPSc types: type 1 with a relative molecular mass of 21 kDa and the primary cleavage site at residue 82 and type 2 with relative molecular mass of 19 kDa and the primary cleavage site at residue 97.20, 21, 28 The two PrPSc types in conjunction with the codon 129 genotype largely explained CJD phenotypic variability and for the first time provided a molecular basis for disease classification (ie, MM1, MM2, VV1, etc).29 More recently, the study of PrPSc under stringent pH conditions using a gel electrophoresis technique with increased sensitivity demonstrated that PrPSc types 1 and 2 are heterogeneous species, which can be further distinguished into molecular subtypes that better fit the current histopathological classification of sporadic CJD (sCJD) into six subtypes.30
Despite these significant advances, much remains to be performed to explore the molecular basis of CJD heterogeneity and strain diversity in full. One intriguing aspect of CJD molecular pathology relates to the co-occurrence of PrPSc types 1 and 2 in the same brain. Originally reported by Parchi et al,29 the existence of CJD subjects with mixed molecular and pathological phenotypes has been confirmed in subsequent studies.31, 32, 33 Nevertheless, difficulties remain in understanding both the biological basis of this phenomenon and its prevalence. The issue has become particularly puzzling after two recent studies34, 35 took advantage of monoclonal antibodies recognizing an epitope between residues 82 and 96 of human PrP located outside the primary PK cleavage site of type 2 PrPSc. These studies showed that all CJD cases classified as type 2, using standard antibodies, also contain a variable amount of PrPSc that matches PrPSc type 1 in gel migration characteristics after PK treatment. It was concluded that all type 2 CJD cases are characterized by the coexistence of PrPSc type 1 and that the two types might always exist in a dynamic equilibrium within the brain. If confirmed, this finding would make the distinction between type 1 and 2 CJD subtypes quantitative rather than qualitative and would question both the validity of the electrophoretic PrPSc mobilities as surrogate markers for prion strains or disease subtype and the rational basis of current CJD classification.
Addressing these issues further, we reasoned that the type 1-like signal might be an effect of a relatively inefficient hydrolysis of PrPSc type 2 rather than ‘bona fide’, subtype-related PrPSc type 1. In line with this hypothesis, our results reveal that by detecting partially cleaved fragments produced by PK digestion in both PrPSc types 1 and 2, the recently developed type 1-selective antibodies easily lead to significant PrPSc typing pitfalls. Moreover, we show here that using a relatively high PK activity the detection sensitivity of type 1 and 2 co-occurrence can be significantly enhanced without the need for ‘type-selective’ antibodies, making this approach the current best for the study of PrPSc types co-occurrence in prion disease.
MATERIALS AND METHODS
Patients and Tissues
We studied 53 sCJD cases and 4 vCJD cases phenotypically characterized in regard to clinical and histopathological features, pattern of PrP deposition, PRNP genotype, and western blot profile of PrPSc. sCJD subtypes were classified according to Parchi et al.28 They included 10 MM1, 5 MV1, 3 VV1, 9 MV2 with kuru plaques, 15 VV2, 3 MM2-cortical, 5 MM2-thalamic, a disease subtype also known as sporadic fatal insomnia,36 and 3 MM sCJD cases in which standard PrPSc typing demonstrated the co-occurrence of types 1 and 2, as described by Parchi et al.29 Brain tissues were obtained at autopsy and were kept frozen at −80°C until use. One sample from the frontal cerebral cortex (FC), usually the middle frontal gyrus (MFG), was available in all cases. In three MV2, three VV2, three MM2-cortical cases, tissues were also obtained from three or more of the following areas: occipital (OCC) or parietal cortex (PC), putamen (PUT), thalamus (TH) and cerebellum (CE). Finally, autopsy brain tissue from three non-demented subjects with a negative neuropathologic examination was used as negative controls.
Antibodies
The following mouse monoclonal antibodies recognizing different human PrP epitopes were used at defined concentrations: 3F4 (residues 108–111)37 obtained from Signet Labs (Dedham, MA, USA) at 80 ng/ml, 12B2 (residues 89–93)38 at 400 ng/ml, Sha31 (residues 145–152) and SAF60 (residues 157–161)39 at 400 and 200 ng/ml, respectively. Antibody concentrations were chosen to yield comparative signal intensities on western blots.
Molecular Genetics
Genomic DNA was extracted from blood or frozen brain tissue. Genotyping of the PRNP coding region was performed as described,28 after receiving an informed consent from the patients or their relatives.
Sample Preparation and Western Blotting
Brain homogenates (10%, wt/vol) were prepared on ice in a lysis buffer with high buffer capacity (LB100) (pH 6.9; 100 mM Tris, 100 mM NaCl, 10 mM EDTA, 0.5% NP-40, 0.5% sodium deoxycholate). Since the pH of Tris buffers changes significantly according to the buffer temperature, the lysis buffers were titrated to pH 6.9 at 37°C (ie, the temperature at which protease digestion is performed). Total protein concentration was estimated using a standard colorimetric method based on bicinchoninic acid (Pierce Biotechnology, Rockford, IL, USA). All samples were diluted at 6 mg protein/ml before protease digestion. If not otherwise specified, aliquots were treated for 1 h at 37°C with PK (47.9 U/mg, Roche) at several concentrations from 0.12 to 256 U/ml (ie, 1 U/ml corresponds to 50 μg/ml when PK-specific activity is 20 U/mg). Since PK generated unspecific bands in the western blots at a concentration higher than 256 U/ml, to reach a complete PrPSc hydrolysis we increased the incubation times rather than PK concentration. Protease digestion was terminated by the addition of 2 mM phenylmethylsulfonyl fluoride. Samples were resuspended in sample buffer (final concentration: 3% sodium dodecyl sulfate (SDS), 4% β-mercaptoethanol, 10% glycerol, 2 mM EDTA, 62.5 mM Tris, pH 6.8) and boiled for 8 min before loading.
In the experiments determining the detection sensitivity of PrPSc type 1 and 2 co-occurrence, frontal cortex homogenates of ‘pure’ PrPSc types 1 and 2 from MM1 and MM2-cortical subjects, respectively, were mixed in ratios of 200:100, 100:100, 70:100, 50:100, 40:100, 30:100, 20:100, 10:100, 5:100, 3:100, 2:100, 1:100, according to their PrPSc content, and digested with 2 or 16 U/ml PK. Similarly, the PK digestion profile of artificially mixed PrPSc types 1 and 2, was determined using frontal cortex homogenates containing 5% of PrPSc type 1 from an MM subject and 95% of PrPSc type 2A from a case showing the MM2-cortical phenotype. To optimize PrPSc co-occurrence, detection samples were concentrated by methanol precipitation and resuspended in a volume equivalent to the one of the original 10% homogenate (ie, 20 μl of sample corresponded to 2 mg of wet tissue).
Protein samples (brain tissue equivalent to 0.2–2 mg of wet tissue) were separated in 12 or 15% Tris/glycine SDS-polyacrylamide gels (37.5:1 acrylamide/bis-acrylamide) using gel electrophoresis apparatus holding running gels of different lengths (5.5 cm for the standard system and 15 cm for the high-resolution system, Bio-Rad). Proteins were transferred to Immobilon P (Millipore) for 2 h at 60 V, blocked with 10% non-fat milk in Tween-Tris-buffered saline, pH 7.5, and probed with the appropriate antibody. The immunoreactivity was visualized by enhanced chemiluminescence (ECL standard, Amersham) on Kodak BioMax Light films (Eastman Kodak).
RESULTS
‘Type 1 Selective’ 12B2 Antibody Detects a Band Migrating Similarly to Type 1 (Type 1-Like) in All Type 2 CJD Subtypes
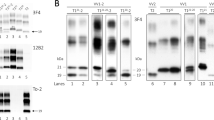
Western blot analyses of PK-digested brain homogenates using 12B2, a monoclonal antibody recognizing an epitope (89–93) located outside PrPSc type 2 primary PK cleavage site (ie, residue 97), detected a band migrating similarly to type 1 (referred to here as type 1-like) in all CJD type 2 subtypes (Figure 1). The amount of the type 1-like band, however, dramatically decreased when PrPSc was digested with a relatively high PK concentration (20 U/ml) (Figure 1). The relative disappearance of the type 1-like band at 20 U/ml of PK was observed in all type 2 CJD subtypes but MV2. Furthermore, in vCJD, the glycoform ratio of this band reproduced that of the corresponding type 2B rather than the one of a typical CJD type 1 (MM1 or VV1) (Figure 1).

The antibody 12B2 detects a type1-like band in all type 2 CJD subtypes. Standard western blot analysis of PrPSc in frontal cortex homogenates from CJD of the subtypes MM1, MM1+2, MM2-cortical (MM2C), vCJD, VV2, MM2-thalamic (MM2T), and MV2. Samples were digested at two different PK concentrations expressed in U/ml (1 U/ml corresponds at 50 μg/ml when specific activity is 20 U/mg; see Materials and Methods for more details) and probed with either 3F4 or 12B2. Approximate molecular masses are in kilodaltons.
A Relatively Inefficient PrPSc Hydrolysis Generates Partially Cleaved Fragments
Given that PK digestion of PrPSc is not a single cleavage step and that the sensitive N-terminal portion of PrPSc has several PK cleavage sites near the resistant core (see PeptideCutter program of the ExPASy Proteomics Server us.expasy.org/tools/peptidecutter) (Figure 2), we explored the possibility that the type 1-like/type 2 co-occurrence detected by selective antibodies such as 12B2 represents, in most cases, an inefficient hydrolysis of type 2 rather than bona fide, CJD subtype or strain-related, type 1. To test this hypothesis, we first analyzed the PrPSc fragments generated by different PK concentrations using a high-resolution gel electrophoresis system (SDS-PAGE 15%, 15 cm long gel) in all CJD subtypes. When exposed to a relatively moderate PK activity (from 0.12 to 0.5 U/ml, for 1 h at 37°C, pH 6.9, 6 mg protein/ml), both types 1 and 2 showed, in addition to the 21 kDa (type 1) or 19 kDa (type 2) band, respectively, a tight group of slower migrating bands (Figure 3) matching the characteristics of partially cleaved fragments of PrPSc theoretically expected based on the availability of PK digestion sites along the PrP sequence. The fact that only the smallest five of these fragments were detected is explained by the co-migration of the larger peptides with the monoglycosylated band of PrP27-30. As expected in a typical step-by-step peptide digestion, a gradual increase in PK concentration significantly reduced the number of these fragments while raising the amount of the resistant core (eg, faster migrating band). Interestingly, when we used a lower resolution electrophoretic apparatus (SDS-PAGE 12%, 5.5 cm long gel) comparable to those commonly used for PrPSc typing, this ladder of fragments appeared as a single band indistinguishable from the resistant core (data not shown). To obtain a better characterization of these peptides, we also performed a western blot analysis using antibodies against different PrP epitopes and a high-resolution gel electrophoresis system. The results showed that, like 3F4 (residues 108–111), SAF60 (residues 157–161) and Sha31 (residues 145–152) antibodies detect all fragments including those corresponding to type 1 and 2 primary cleavage sites (ie, cleaved at 82 and 97), whereas 12B2 (residues 89–93) did not detect the 19 kDa fragment corresponding to the PrPSc type 2 core. Moreover, this antibody only weakly recognized the fragment of about 20 kDa, migrating just above the PrPSc type 2 core, which is in line with the fact that the corresponding theoretically expected fragment lacks one residue in the 12B2 epitope (Figure 4).

Diagram of human PrPSc showing the primary cleavage sites for both types 1 and 2 resistant cores, and the theoretical expected PK cleavage sites in their corresponding N-terminal PK-sensitive portions (obtained by the PeptideCutter program of the ExPASy Proteomics Server; us.expasy.org/tools/peptidecutter). Primary cleavage by PK at each of these sites generates partially cleaved fragments. The fragments we could identify in both types 1 and 2 are marked with v. The epitopes recognized by the monoclonal antibodies 3F4 and 12B2 are also marked.

Effect of PK concentration on PrPSc digestion profiles. Western blot analysis of PrPSc types 1 (MM1 subtype) and 2 (VV2 subtype) probed with 3F4 antibody after digestion at different PK concentrations. A high-resolution gel electrophoresis system (SDS-PAGE 15%, 15 cm long gel) was used. In the lower panel the band with an approximate MW of 21 kDa is marked with an asterisk. PK activity is indicated as either U/ml or μg/ml (1 U/ml corresponds at 50 μg/ml when specific activity is 20 U/mg). The same result was reproduced twice with samples from at least four subjects of each group. Approximate molecular masses are in kilodaltons.

Detection of partially cleaved fragments with different antibodies. Western blot analysis of PrPSc types 1 (MM1 subtype) and 2 (VV2 subtype) after digestion with 0.25 U/ml of PK, using different antibodies (epitopes are shown). A high-resolution gel electrophoresis system (SDS-PAGE 15%, 15 cm long gel) was used. After blotting, the PVDF membrane was cut into four parts that were incubated with different antibodies and then reassembled for the final film exposure. Approximate molecular masses are in kilodaltons.
PK Digestion Profiles of Type 1-Like and Partially Cleaved PrPSc Fragments Differ from that of PrP27-30
To further explore the possibility that the partially cleaved fragments fully constitute the type 1-like signal detected by 12B2, we performed, on all sCJD subtypes as well as vCJD, a set of experiments in strictly controlled, homogeneous digestion conditions, using several PK concentrations, a high-resolution gel electrophoresis system, and both 3F4 and 12B2 monoclonal antibodies (Figure 5). We found that PrPSc digestion in the presence of a relatively low PK activity (0.5–2 U/ml) produces a decrease in the full-length form, which appears to be threefold more PK resistant than the PrPC from CJD-negative cases (data not shown), and a gradual increase in both PrPSc resistant core and partially cleaved fragments. At increasing PK concentrations, however, the total amount of partially cleaved fragments begins to decrease while PrPSc resistant core continues to increase until the PK activity becomes 8–32 times higher, depending on the CJD subtype.

PK digestion profiles of PrPSc in all sCJD subtypes and vCJD: comparison between 3F4 and 12B2 antibodies. (a) Western blot profiles of PrPSc from MM1 and MM2-cortical (MM2C) subtypes digested at different increasing PK concentrations and probed with either 3F4 or the ‘type 1-selective’ 12B2. A high-resolution gel electrophoresis system (SDS-PAGE 15%, 15 cm long gel) was used. Windows correspond to higher exposure times. Approximate molecular masses are in kilodaltons. (b) Summary of results for all CJD subtypes. Values refer to the amount of different PrPSc forms and are expressed in arbitrary units calculated by densitometric analyses of chemiluminescence western blot signals generated by unglycosylated PrPSc in duplicate blots probed with either 3F4 or 12B2 antibodies. Data are mean±s.d. (n=3). The profiles of full-length PrP (—), PrPSc core resistant fragment (–▴–), PrPSc partially cleaved fragments (–▵–), and sum of all PrPSc fragments (—) were obtained from the blots probed with 3F4, whereas the profile of type 1-like PrPSc (–○–) was obtained from the blots probed with 12B2. The profile of full-length PrP obtained with 12B2 overlapped with that obtained with 3F4 and was omitted. PK activity is expressed in U/ml (1 U/ml corresponds at 50 μg/ml when specific activity is 20 U/mg). Protease digestion was performed at 37°C for 1 h at all PK concentrations, but the last three points (marked with #) where the digestion was performed at 37°C for different times at the highest PK concentration (ie, 256 U/ml). The short arrow (↓) indicates the highest value obtained for the sum of all fragments, whereas the double arrow (↕) corresponds to the highest difference between the value of PrP27-30 and that of partially cleaved fragments.
Using 12B2 at a concentration in which the antibody shows a similar affinity for PrP as 3F4, we compared the PK digestion profiles of the type 1-like signal disclosed by 12B2 in CJD type 2 cases with those of the PrP bands detected by 3F4 in all CJD subtypes. The data show that the type 1-like signal detected by 12B2 in CJD type 2 cases is significantly less resistant to PK digestion than PrPSc resistant core from either MM1 or VV1 subtypes, while largely matches that of the partially cleaved fragments detected by 3F4 (Figure 5b). More precisely, the type 1-like signal appears to be even more PK sensitive than the partially cleaved fragments detected by 3F4 (Figure 5b), but this can be explained by the fact that 12B2, due to his affinity properties, only weakly recognizes the 20 kDa band, which corresponds to the smallest of the type 2-associated partially digested fragments (Figure 4). Similar results were obtained in each of the CJD type 2 subtypes coupled with a homozygous codon 129 genotype. Furthermore, the same results were also reproduced in three different areas (ie, frontal cortex, putamen, and thalamus) of six sCJD cases (two MM1, two VV2, and two MM2-cortical) (data not shown). Overlapping digestion profiles between type 1-like and type 2 signals were only obtained in samples from the MV2 subtype (Figure 5b), where, however, the type 1 signal was also clearly visible with 3F4 (Figure 1), a finding in full agreement with our previous observation28, 29 that the PrPSc core in sCJDMV2 comprises a doublet in most cases. Interestingly, the analyses of the PrPSc digestion profiles (Figure 5b) showed that the maximal density of the PrPSc band (Dmax), corresponding to the sum of all fragments and therefore optimal for prion detection, was achieved with PK concentrations from 1 to 8 U/ml (corresponding to 50–400 μg/ml when specific PK activity is 20 U/mg) depending on the subtypes, whereas the highest PrPSc resistant core detection, with only traces of associated partially digested fragments that are optimal for typing analysis (Topt), was reached with PK concentrations from 8 to 32 U/ml. Thus, experimental conditions that are ideal for the most sensitive PrPSc detection do not coincide with those, which are best for PrPSc typing.
To further compare the PK digestion profiles of type 1-like and bona fide type 1, we artificially mixed PrPSc types 1 and 2 in a 5% (MM1)/95% (MM2-cortical) ratio. Despite its minor proportion, type 1 showed a distinguishable band also at the highest PK concentration used (256 U/ml) making its profile clearly distinct from that of type 1-like (Figures 5a and 6).

PK digestion profiles of PrPSc in artificially mixed sCJD subtypes. Western blot profiles of PrPSc in artificially mixed sCJD subtypes with little amount of PrPSc type 1 (5% MM1 added to 95% MM2-cortical). Protease digestion was performed at different increasing PK concentrations; membranes were probed with 3F4 and the ‘type 1-selective’ antibody, 12B2. A high-resolution gel electrophoresis system (SDS-PAGE 15%, 15 cm long gel) was used. The same result was reproduced with samples from at least three subjects. Approximate molecular masses are in kilodaltons.
Regional Variations of PrPSc Type 1 and 2 Co-Occurrence
The analyses of sCJD cases with real type 1 and 2 co-occurrence revealed by the 3F4 antibody show significant regional differences in the amount ratio of the two protein types (Figure 7a), correlating with the pathological disease phenotype (data not shown).29, 31 To see whether similar regional variations are observed in the type 1-like and type 2 ratio, we analyzed different areas of type 2 sCJD cases after PK digestion in standardized and homogenous conditions. In particular, much attention was given to performing PK digestion of tissue homogenate at a given pH value (using a high buffer capacity solution) and at a given total protein concentration. This is because PrPSc digestion by PK is significantly affected by both the actual pH30 and the total protein concentration in the homogenate, which can also significantly differ among different areas (data not shown). In these conditions, the amount ratio between the type 1-like band and type 2 signal was not significantly influenced by the brain structure analyzed (Figure 7a). Again, the only exception was represented by the MV2 cases (Figure 7b). However, as already stated, the co-occurrence of bands in MV2 is already and even more readily detected by the 3F4 antibody.

Regional western blot profiles of PrP27-30 from different sCJD subtypes. Standard western blot analysis of PrP27-30 obtained from (a) different brain areas of VV2, MM2, MM1+2 cases. Samples were digested with 2 U/ml PK and probed with either 3F4 or the ‘type 1-selective’ antibody, 12B2. Similar results were obtained with samples from at least four subjects. (b) Different brain areas of a MV2 case sample was digested with two different PK concentrations and probed with either 3F4 or the type 1-selective antibody, 12B2. Similar significant differences were obtained with samples from at least three subjects. Approximate molecular masses are in kilodaltons. For brain areas abbreviations see Materials and Methods.
Detection Sensitivity of PrPSc Type 1 and 2 Co-Occurrence in the Same Sample by 3F4
To measure the limit of detection of type 1 and 2 co-occurrence by 3F4 antibody, we mixed two homogenates containing pure type 1 or 2 PrPSc from MM1 and MM2-cortical subjects, in various proportions. We then analyzed the samples by immunoblotting (3F4 antibody) after digestion with either 2 U/ml, the protease concentration most commonly used for PrPSc detection and typing, or 16 U/ml of PK, the average of the protease concentrations at which we obtained the highest PrPSc resistant core detection with only traces of associated partially digested fragments (Topt) in the different CJD subtypes (Figure 5b). Using a 16 U/ml PK concentration, the detection threshold for type 1 was 9% of total PrPSc while it was 33–41% of total PrPSc using the 2 U/ml PK concentration (Figure 8a). In contrast, the detection of low amounts of type 2 in the presence of a relatively high amount of type 1 was not significantly affected by PK concentration/activity. Furthermore, by using a high-resolution gel electrophoresis system (ie, 15% Tris-glycine SDS-PAGE, 15 cm long) combined with a higher sample concentration, it was possible to raise the detection threshold for type 1 or type 2 up to a limit of 3–5% of total PrPSc (Figure 8b).

Detection limit of PrPSc types 1 and 2 co-occurrence by 3F4. Two brain homogenates containing either pure type 1 or pure type 2 were mixed in various ratios, as indicated. Two aliquots of each sample were digested using different PK concentrations. To optimize detection of PrPSc co-occurrence, the samples were concentrated fourfold by methanol precipitation. Western blot analysis was performed by using both (a) conventional gel electrophoresis system (SDS-PAGE 12%, 5.5 cm long gel) and (b) high-resolution gel electrophoresis system (SDS-PAGE 15%, 15 cm long gel). The antibody 3F4 was used. The same result was reproduced twice with samples from at least three subjects of each group. Approximate molecular masses are in kilodaltons.
DISCUSSION
In the absence of a disease-specific nucleic acid, the molecular diagnosis of prion diseases must rely on protein studies. Besides PrPSc detection, which is required for a definite diagnosis of prion disease, the study of PrPSc physicochemical properties has assumed crucial importance for both strain typing and molecular classification of TSEs. Thus, refined molecular techniques in prion diagnostics should aim to improve not only PrP detection sensitivity but also discriminatory properties among abnormal PrP isoforms. Difficulties in current CJD postmortem diagnostics relate to the finding of PrPSc types coexistence in the brains. CJD cases with mixed molecular and pathological features have been reported in several studies,29, 31, 32, 33 but no criteria have been established for the correct biochemical biologically relevant identification of PrPSc types co-occurrences. Furthermore, it is still unclear to what extent current molecular techniques allow the detection of the whole spectrum of such cases. In this regard, antibodies recognizing an epitope between residues 82 and 96 (ie, not detecting type 2) appeared straightforward tools to discriminate cases of animal-derived TSE strains.38, 40, 41, 42, 43 Using these antibodies on human CJD isolates, two groups of investigators recently found that all type 2 samples show at least some associated protein migrating similarly to PrPSc type 1 and reached the conclusion that PrPSc type 1 regularly coexist in CJD previously classified as type 2.34, 35 We also detected a type 1-like band in all CJD type 2 subtypes but came up to a different conclusion concerning the origin and biological significance of this band.
In a previous study,30 where we originally applied a high-resolution electrophoretic system, which better discriminates between distinct PrP isoforms, we showed that PK digestion of both PrPSc types 1 and 2 produces additional fragments migrating slightly slower than the resistant core. Since these fragments were significantly more sensitive to protease digestion than the PrPSc core, we thought they might represent the effect of a relatively inefficient hydrolysis of PrPSc. Whereas the type 1-associated fragments do not interfere with type 2 detection, it is noteworthy that the size of associated fragments generated by PrPSc type 2 protease digestion overlaps with that of PrPSc type 1. Furthermore, this ladder of peptides is visualized as a single band in a conventional 5.5 cm gel system, giving the misleading impression that they represent a PrP27-30 type 1 core fragment. The present study reanalyzed these peptides using a high-resolution gel electrophoretic system and both non-discriminatory and type 1-selective antibodies. We calculated that the observed fragments exactly match the number and size of the theoretically expected partially cleaved PrPSc fragment generated by PK digestion. By analyzing the degree of PK resistance of these peptides compared to that of PrPSc type 1 and 2 cores in greater detail (Figure 5), we showed that the band detected by 12B2 in each CJD type 2 subtype but the MV2, is not PrPSc type 1, but rather a band that correspond to the above-mentioned associated fragments. The PK resistance profile of this band, in fact, was similar to that of partially cleaved fragments detected by 3F4, but significantly different from that of the PrPSc type 1 resistant core from either MM1 or VV1. Yull et al35 addressed this issue performing a PK-digestion time–course experiment in vCJD brain. They observed a rapid initial decline in the signal detected by 12B2, which plateaued around 60 min and then apparently remained constant over the next 2 h. According to our data, however, the limited range of PK activities they tested may be the explanation for the apparent plateau effect observed.
It is well known that the glycoform ratio is another property distinguishing between PrPSc isoforms associated with distinct CJD subtypes or prion strains.17, 19, 29, 44 Yull et al35 found that the type 1-like band detected by 12B2 in vCJD maintained the very characteristic glycoform ratio of PrPSc type 2B and concluded that the band they considered a PrPSc resistant core could not be a common type 1. We consider this result, which was also reproduced by us, as further evidence that the type 1-like band recognized by 12B2 in vCJD cases represents partially cleaved fragments of PrPSc type 2B rather than real, bona fide PrPSc type 1.
We agree with Yull et al35 and Polymenidou et al34 that the detection of type 1 and 2 co-occurrence using a conventional typing method and antibodies recognizing both types may lack sufficient sensitivity. In this regard, we were able to increase the sensitivity of the type 1 and 2 co-occurrence signal up to the value of 10% (weak band of one type/total PrPSc) using a conventional gel electrophoresis system and up to 3–5% using a high-resolution gel electrophoresis system. This result was achieved by increasing the PK concentration to 16 U/ml (Topt), hence by reducing the amount of partially cleaved fragments the specific resistant core could be detected more distinctively. In their study, Polymenidou et al34 suggested that the use of a too high PK concentration could lead to false-negative results and that the PK concentration for the most reliable PrPSc detection should be the minimum concentration that will completely digest PrPC. We agree that the highest sensitivity is needed when the primary goal is a positive or negative diagnosis based on PrPSc detection; however, at present, this is rarely a significant issue in CJD diagnosis given the relatively high PrPSc concentration in CJD brains, the only exceptions being rare cases of genetic TSEs such as FFI or familial CJD associated with extra-repeat insertions44 (unpublished data). However, when a fine typing analysis is needed, the optimum PK concentration corresponds to the one generating the minimum amount of partially digested fragments together with the maximum possible amount of PrP27-30 resistant core. This study shows that this condition is reached at a PK concentration of 16 U/ml (Topt). Given that the decrease of PrPSc signal in this condition compared to the maximal detection sensitivity, obtained at 2 U/ml PK (Dmax), was never higher than 30%, it is possible to overcome this decrease by concentrating the sample up to fourfold, using a rapid method of protein precipitation (eg, methanol precipitation).
Additional data concerning the regional variability of PrPSc physicochemical properties within the same brain further support our conclusions. It is well known that CJD subtypes significantly differ in the intracerebral distribution of PrPSc.21 This is related to the existence of different prion strains and, to a lesser extent, to the host genotype variability such as that determined by the codon 129 polymorphism. Thus, in a CJD case with PrPSc co-occurrence, it should be expected that the relative amount of the two proteins varies significantly according to the brain area analyzed (Figure 8). This is exactly what we found in a recent study where the regional properties of PrPSc in about 20 different brain structures of 40 CJD cases with PrPSc types co-occurrence were analyzed by western blotting (P Parchi et al, unpublished data). In contrast, the present study detected a constant amount of type 1-like/type 2 ratio in different areas of the same case in all type 2 CJD subtypes (ie, MM2-cortical, MM2-thalamic, VV2, vCJD) but the MV2. It must be also emphasized that both studies applied a homogeneous PK digestion condition. In particular, much attention was given to protein concentration and the actual homogenate pH, two experimental variables that may vary significantly among brain regions and significantly affect PK activity.
The findings in the MV2 sCJD subtype deserve further comment. The western blot profile of PrP27-30 in these cases appears almost invariably characterized by the association of two PrPSc core fragments, which include a classic 19 kDa type 2 band and a slower migrating band of about 20 kDa. As previously reported by us,29, 30 the two bands are readily detected by 3F4. Furthermore, their relative amount varies significantly according to the brain region analyzed, although the type 2 band is usually dominant over the 20 kDa band (P Parchi et al, unpublished data). Thus, the association truly reflects a co-occurrence of PrPSc types. We understand that it is tempting to call this PrPSc pattern a type 1 and 2 association, but we feel this would be incorrect for the following reasons: (i) the 20 kDa band shows a slightly faster migration and a quite distinct regional distribution than the classic type 1 protein found in MM and MV cases; (ii) although a band with similar migration pattern is seen in VV1 cases, again, the regional distribution of the protein fragment in the two sCJD subtypes (ie, VV1 and MV2) is quite distinct (P Parchi et al, unpublished data); (iii) there are MV sCJD cases with a PrPSc type 1 and 2 co-occurrence (ie, real 21+19 kDa bands) showing a quite distinct pathological phenotype from that of the MV2 cases with kuru plaques. Thus, an alternative nomenclature should be applied to this PrPSc profile. For the moment, we propose defining the pattern as type 2 associated with an upper band of 20 kDa (ie, PrPSc type 2+20 kDa).
In conclusion, the recently developed type 1-selective antibodies (ie, antibodies recognizing an epitope between the type 1 and 2 N-termini of human CJD cases) are of potential interest for the study of the physicochemical properties of a type 1 component, but only once the co-occurrence of PrPSc types has been confirmed with standard antibodies. Indeed, the possibility to maximize the detection of 21 kDa PrP band and the partially cleaved fragments in the absence of the signal generated by the 19 kDa type 2 band easily leads to an incorrect interpretation of the type-related nature of the detected band. This is particularly true when a conventional gel electrophoretic system is used where different peptides appear as a single band with a PrPSc type 1 migration pattern. On the other hand, setting up an experimental condition where a pure type 2 signal without any trace of associated PrP-type 1-like band is obtained is virtually unthinkable, given that the core fragment itself is only partially resistant to protease digestion.
Beside, we demonstrated that combining high-resolution gel electrophoresis and a relatively high PK activity and protein loading, the detection sensitivity of type 1 and 2 co-occurrence can be increased 10-fold without the need for ‘type-selective’ antibodies, making it the current best approach for the detection of PrPSc type co-occurrence in human prion disease. It will now be important to apply this refined western blot approach to a systematic analysis of several brain regions in a large cohort of CJD cases to reach a reliable estimate of the extent of PrPSc type co-occurrence in CJD. Nevertheless, our current data already show that there are many type 2 CJD cases lacking evidence of the co-occurrence of bona fide PrPSc type 1 in several brain regions, indicating that PrPSc type 1 coexistence in type 2 CJD cases is not the rule.
References
Prusiner SB . Prions. Proc Natl Acad Sci USA 1998;95:13363–13383.
Büeler H, Aguzzi A, Sailer A, et al. Mice devoid of PrP are resistant to scrapie. Cell 1993;73:1339–1347.
Caughey BW, Dong A, Bhat KS, et al. Secondary structure analysis of the scrapie-associated protein PrP27-30 in water by infrared spectroscopy. Biochemistry 1991;30:7672–7680.
Pan KM, Baldwin M, Nguyen J, et al. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc Natl Acad Sci USA 1993;90:10962–10966.
Safar J, Cohen FE, Prusiner SB . Quantitative traits of prion strains are enciphered in the conformation of the prion protein. Arch Virol Suppl 2000;16:227–235.
Bolton DC, McKinley MP, Prusiner SB . Identification of a protein that purifies with the scrapie prion. Science 1982;218:1309–1311.
Gambetti P, Kong Q, Zou W, et al. Sporadic and familial CJD: classification and characterisation. Br Med Bull 2003;66:213–239.
Kretzschmar HA, Parchi P . Pathology and genetics of human prion diseases. In: Hoernlimann B, Riesner D, Kretzschmar H (eds). Prions in Humans and Animals. De Gruyter: Berlin/New York, 2007, pp 287–305.
Bruce ME, McConnell I, Fraser H, et al. The disease characteristics of different strains of scrapie in Sinc congenic mouse lines: implications for the nature of the agent and host control of pathogenesis. J Gen Virol 1991;72:595–603.
Palmer MS, Dryden AJ, Hughes JT, et al. Homozygous prion protein genotype predisposes to sporadic Creutzfeldt–Jakob disease. Nature 1991;352:340–342.
Kong Q, Surewicz WK, Petersen RB, et al. Inherited prion diseases. In: Prusiner SB (ed). Prion Biology and Diseases, 2nd edn. Cold Spring Harbor: New York, 2004, pp 673–775.
Barron RM, Thomson V, Jamieson E, et al. Changing a single amino acid in the N-terminus of murine PrP alters TSE incubation time across three species barriers. EMBO J 2001;20:5070–5078.
Telling GC, Parchi P, DeArmond SJ, et al. Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science 1996;274:2079–2082.
Bruce ME, Will RG, Ironside JW, et al. Transmissions to mice indicate that ‘new variant’ CJD is caused by the BSE agent. Nature 1997;389:498–501.
Parchi P, Brown P, Capellari P, et al. Agent strain variation in human prion diseases: insights from transmission to primates. In: Iqbal K, Swaab DF, Winblad D, Wisniewski HM (eds). Alzheimer' s Disease and Related Disorders: Etiology, Pathogenesis, and Therapeutics. Wiley J & Sons Ltd: Chichester, UK, 1999, pp 561–567.
Korth C, Kaneko K, Groth D, et al. Abbreviated incubation times for human prions in mice expressing a chimeric mouse–human prion protein transgene. Proc Natl Acad Sci USA 2003;100:4784–4789.
Kascsak RJ, Rubenstein R, Merz PA, et al. Immunological comparison of scrapie-associated fibrils isolated from animals infected with four different scrapie strains. J Virol 1986;59:676–683.
Bessen RA, Marsh RF . Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. J Virol 1994;68:7859–7868.
Monari L, Chen SG, Brown P, et al. Fatal familial insomnia and familial Creutzfeldt–Jakob disease: different prion proteins determined by a DNA polymorphism. Proc Natl Acad Sci USA 1994;91:2839–2842.
Parchi P, Castellani R, Capellari S, et al. Molecular basis of phenotypic variability in sporadic Creutzfeldt–Jakob disease. Ann Neurol 1996;39:767–778.
Parchi P, Capellari S, Chen SG, et al. Typing prion isoforms. Nature 1997;386:232–234.
Collinge J, Sidle KCL, Meads J, et al. Molecular analysis of prion strain variation and the aetiology of ‘new variant’ CJD. Nature 1996;383:685–690.
Somerville RA, Chong A, Mulqueen OU, et al. Biochemical typing of scrapie strains. Nature 1997;386:564.
Safar J, Wille H, Itri V, et al. Eight prion strains have PrPSc molecules with different conformations. Nat Med 1998;4:1157–1165.
Caughey B, Raymond GJ, Bessen RA . Strain-dependent differences in beta-sheet conformations of abnormal prion protein. J Biol Chem 1998;273:32230–32235.
Parchi P, Notari S, Stramiello R, et al. History and state of the art of PrP-res ‘typing’ in Creutzfeldt–Jakob disease. In: Kitamoto T (ed). Prions: Food and Drug Safety. Springer-Verlag: Tokyo, 2005, pp 77–96.
Baron T, Biacabe AG, Arsac JN, et al. Atypical transmissible spongiform encephalopathies (TSEs) in ruminants. Vaccine 2007;25:5625–5630.
Parchi P, Zou W, Wang W, et al. Genetic influence on the structural variations of the abnormal prion protein. Proc Natl Acad Sci USA 2000;97:10168–10172.
Parchi P, Giese A, Capellari S, et al. Classification of sporadic Creutzfeldt–Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 1999;46:224–233.
Notari S, Capellari S, Giese A, et al. Effects of different experimental conditions on the PrPSc core generated by protease digestion. J Biol Chem 2004;279:16797–16804.
Puoti G, Giaccone G, Rossi G, et al. Sporadic Creutzfeldt Jakob disease: co-occurrence of different types of PrPSc in the same brain. Neurology 1999;53:2173–2176.
Head MW, Bunn TJ, Bishop MT, et al. Prion protein heterogeneity in sporadic but not variant Creutzfeldt–Jakob disease: UK cases 1991–2002. Ann Neurol 2004;55:851–859.
Schoch G, Seeger H, Bogousslavsky J, et al. Analysis of prion strains by PrPSc profiling in sporadic Creutzfeldt–Jakob disease. PLoS Med 2006;3:e14.
Polymenidou M, Stoeck K, Glatzel M, et al. Coexistence of multiple PrPSc types in individuals with Creutzfeldt–Jakob disease. Lancet Neurol 2005;4:805–814.
Yull HM, Ritchie DL, Langeveld JP, et al. Detection of type 1 prion protein in variant Creutzfeldt–Jakob disease. Am J Pathol 2006;168:151–157.
Parchi P, Capellari S, Chin S, et al. A sporadic prion disease mimicking fatal familial insomnia. Neurology 1999;52:1757–1763.
Kascsak RJ, Rubenstein R, Merz PA, et al. Mouse polyclonal and monoclonal antibody to scrapie-associated fibril proteins. J Virol 1987;61:3688–3693.
Langeveld JPM, Jacobs JG, Erkens JH, et al. Rapid and discriminatory diagnosis of scrapie and BSE in retro-pharyngeal lymph nodes of sheep. BMC Vet Res 2006;2:19.
Feraudet C, Morel N, Simon S, et al. Screening of 145 anti-PrP monoclonal antibodies for their capacity to inhibit PrPSc replication in infected cells. J Biol Chem 2005;280:11247–11258.
Stack MJ, Chaplin MJ, Clark J . Differentiation of prion protein glycoforms from naturally occurring sheep scrapie, sheep-passaged scrapie strains (CH1641 and SSBP1), bovine spongiform encephalopathy (9BSE) cases and Romney and Cheviot breed sheep experimentally inoculated with BSE using two monoclonal antibodies. Acta Neuropathol (Berl.) 2002;104:279–286.
Thuring CMA, Erkens JHF, Jacobs JG, et al. Discrimination between scrapie and bovine spongiform encephalopathy in sheep by molecular size, immunoreactivity, and glycoprofile of prion protein. J Clin Microbiol 2004;42:972–980.
Baron T, Biacabe AG, Bencsik A, et al. Transmission of new bovine prion to mice. Emerg Infect Dis 2006;12:1125–1128.
Espinosa JC, Andréoletti O, Castilla J, et al. Sheep-passaged BSE agent exhibits altered patho-biological properties in bovine-PrP transgenic mice. J Virol 2007;81:835–843.
Parchi P, Castellani R, Cortelli P, et al. Regional distribution of protease-resistant prion protein in fatal familial insomnia. Ann Neurol 1995;38:21–29.
Acknowledgements
We thank Barbara Polischi for her technical support. James Ironside at the National CJD Surveillance Unit in the UK kindly provided the tissues of variant CJD used for this study. SAF60 and Sha31 antibodies were a kind gift from Jacques Grassi at Service de Pharmacologie et d́Immunologie, CEA/Saclay, France. This study was supported by the European Commission (FOOD-CT-2004-506579) and the Italian Ministry of University, Research and Technology (FIRB-2003-RBNE03FMCJ_006).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Notari, S., Capellari, S., Langeveld, J. et al. A refined method for molecular typing reveals that co-occurrence of PrPSc types in Creutzfeldt–Jakob disease is not the rule. Lab Invest 87, 1103–1112 (2007). https://doi.org/10.1038/labinvest.3700676
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.3700676
Keywords
This article is cited by
-
Environmental and host factors that contribute to prion strain evolution
Acta Neuropathologica (2021)
-
Co-existence of PrPD types 1 and 2 in sporadic Creutzfeldt-Jakob disease of the VV subgroup: phenotypic and prion protein characteristics
Scientific Reports (2020)
-
Co-occurrence of chronic traumatic encephalopathy and prion disease
Acta Neuropathologica Communications (2018)
-
Distinct pathological phenotypes of Creutzfeldt-Jakob disease in recipients of prion-contaminated growth hormone
Acta Neuropathologica Communications (2015)
-
Deciphering the pathogenesis of sporadic Creutzfeldt-Jakob disease with codon 129 M/V and type 2 abnormal prion protein
Acta Neuropathologica Communications (2013)
