
Abstract
Telomeres are ribonucleoprotein structures that protect the end of linear chromosomes from recognition as DNA double-stranded breaks and activation of a DNA damage response. Telomere-associated proteins also regulate telomerase, the protein responsible for maintaining telomere length. Loss of telomere function results from either alteration in the capping function at telomeres, or from progressive loss of telomeric repeats necessary to maintain proper telomeric structure. Dysfunctional telomeres activate p53 to initiate cellular senescence or apoptosis to suppress tumorigenesis. However, in the absence of p53, telomere dysfunction is an important mechanism to generate chromosomal instability commonly found in human carcinomas. Telomerase is expressed in the majority of human cancers, making it an attractive therapeutic target. Emerging anti-telomerase therapies that are currently in clinical trials might prove useful against some forms of human cancers.
Similar content being viewed by others
Main
The development of human carcinomas is intimately linked to advancing age, with the vast majority of cancers occurring in older adults. Adult cancers are predominantly carcinomas of epithelial origin, arising in compartments that undergo continual renewal throughout life. For example, such cycles of proliferation and replacement are especially prominent in the breast where mammary secretory epithelial cells undergo hormonally regulated proliferation and regression coupled to each menstrual cycle. A feature of human carcinomas is their strikingly complex cytogenetic profiles, characterized by the presence of complex non-reciprocal translocations (NRTs).1 NRTs result from random and improper fusions of broken chromosomes leading to gains or losses of chromosomal segments, some of which likely impinge upon cancer-relevant pathways. Indeed, the vast majority of sporadic breast carcinomas display chromosomal instability (CIN) and are called CIN neoplasias.2, 3 While the molecular mechanisms underlying CIN are not well understood, it likely involves disruption of multiple genes with ‘caretaker’ functions.4, 5 Loss of function of tumor suppressor genes, including p53, is associated with increased genomic instability. However, mutations in p53 usually occur late during tumor progression, well after they have acquired significant genomic instability.6, 7
Importance of telomere end protection
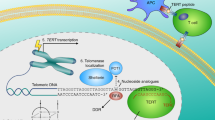
One important mechanism that can give rise to CIN is the functional status of telomeres. Telomeres are composed of TTAGGG repeats that associate with telomere-specific binding proteins.8 The synthesis and maintenance of telomeric repeats are mediated by telomerase, a specialized ribonucleoprotein complex composed of a RNA component (Terc) and its protein counterpart (Tert). In the absence of telomerase, the failure of DNA polymerase to fully synthesize terminal ends of the lagging DNA strand leads to progressive telomere shortening with each round of replication. In human somatic tissues, the strict downregulation of telomerase accounts for the age-dependent decline in telomere lengths in somatic cells.9, 10 These studies have documented a decrease in telomere length in several human epithelial cell types, ranging from 50 to 100 bp per population doubling, for a total lifetime loss of approximately 2–4 kb. This rate of telomere length attrition would be significant in long-lived organisms such as humans.
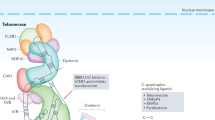
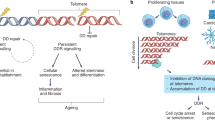
Telomeres serve at least three essential functions: (1) protecting natural chromosomal DNA ends from being inappropriately recognized as double-stranded breaks (DSBs) and therefore initiating an inappropriate DNA damage response (DDR), (2) protecting chromosomal ends from inappropriate enzymatic degradation and (3) preventing chromosomal end-to-end fusions. Three sequence specific DNA binding proteins are recruited to chromosomal ends: the duplex telomere-binding proteins TRF1 and TRF2 and the single-stranded TTAGGG repeat binding protein POT1. These proteins are interconnected by three additional proteins: TIN2, TPP1 and RAP1 to form a functional complex that caps telomeric ends and prevent inappropriate activation of non-homologous end joining (NHEJ) and homologous recombination (HR) pathways at telomeres (Figure 1a). The notion that telomere-binding proteins protect chromosomal ends from being recognized as DSBs is supported by observations that deletion of components of this complex leads to telomere dysfunction that then engages canonical DDR pathways. The DDR can be envisioned as an intracellular signal-transduction pathway. DNA stimuli are detected by sensor proteins such as MRE11/Nbs1/Rad50 that then trigger the activation of a signal transduction system composed of upstream and downstream protein kinases such as ATM/ATR, Chk1 and Chk2. The DNA damage signal ultimately activates p53, eliciting two potent tumor suppressive activities: apoptosis and cellular senescence that together serve to inhibit further growth of genetically damaged cells (Figure 1b).

(a) Telomere structure. The telomere is postulated form a lariat-like structure, in which the 3′ G-overhang of the telomere invades a duplex telomeric region to form a double-stranded t-loop and a single-stranded D-loop. The six-component shelterin complex is postulated to bind and stabilize this t-loop structure when cells are not replicating. During DNA replication, the 3′ overhang becomes accessible to telomerase to elongate telomeres. Telomere extension by telomerase requires translocation of the POT1 protein to an internal site, permitting telomerase to access the terminal G-residue. The shelterin complex protects telomeres beginning at the G2 phase of the cell cycle from inappropriate non-homologous end joining (NHEJ) and homologous recombination (HR)-mediated repair of telomeric DNA. The six-component core protein complex is shown schematically. (b) Telomere dysfunction elicits a DNA damage response at telomeres and activation of ATM/ATR kinases, resulting in p53-mediated cell-cycle arrest/replicative senescence or apoptosis to suppress further growth of genomically unstable cells. (c) Speculative model, based on available evidence, of how telomere dysfunction initiates genomic changes to promote the development of breast cancer. Loss of the p53-dependent DNA damage checkpoint is postulated to be important for tumor progression. Transition from normal ductal breast epithelium to invasive carcinoma correlates with the presence of dysfunctional telomeres and loss of p53.
Telomere dysfunction-induced cellular senescence as a tumor suppressor mechanism
Primary human cells do not express telomerase and their replicative potential are limited by progressive telomere shortening, eventually resulting in the onset of cellular senescence, a state of permanent cell-cycle arrest in which cells exhibit changes in gene expression, display characteristic morphological changes and stain for senescence-associated β-galactosidase (SA-β-gal) activity.9, 10, 11, 12, 13 Other stresses, such as oncogene activation, can also trigger cellular senescence. Regardless of the source of cellular stress, it appears that generation of damaged DNA is essential to engender the senescence response (Figure 1b).14, 15
Owing to its antiproliferative effects, cellular senescence induced by shortened telomeres has long been postulated to be a potent tumor suppression mechanism in vivo. However, since the vast majority of what is known about cellular senescence comes from cell culture studies, its existence in vivo has been controversial until recently. Only with the advent of genetically engineered mouse models in which telomerase could be experimentally eliminated could one address the interplay between dysfunctional telomeres and the activation of p53-dependent tumor suppressor programs in vivo.16 For example, the INK4a−/− mouse lacks both p16INK4a and p19ARF, which have important roles in both the pRb and p53 pathways. Importantly, the p53-dependent DDR is intact in the INK4a−/− mouse. Treatment of telomerase null, INK4a−/− compound mutant mice possessing long (functional) telomeres with chemical carcinogens revealed that these mice are highly cancer prone.17 However, similar treatment of telomerase null INK4a−/− mice possessing dysfunctional telomeres yielded a reduction in tumor incidence and much longer survival. A similar finding was observed in a skin carcinogenesis model, in which telomerase null mice with dysfunctional telomeres produced significantly fewer advanced skin tumors after chemical carcinogenesis treatment of the skin compared to wild-type control animals with long telomeres.18 Increased p53 staining was readily detected in papillomas possessing dysfunctional telomeres, suggesting that further progression to more advanced skin malignancies was likely inhibited by p53 activation. Because dysfunctional telomeres readily activate an apoptotic program in vivo, it was long thought that dysfunctional telomere-induced apoptosis was the primary mechanism for tumor suppression.
However, given that the aforementioned mouse models all possess wild-type p53 capable of inducing both apoptosis and cellular senescence, it remained unclear how dysfunctional telomeres limit neoplastic growth in vivo. Is p53-dependent cellular senescence important for dysfunctional telomere-dependent tumor suppression? The generation of mutant mice with dysfunctional telomeres in the setting of a p53 knock-in mutation (p53R172P allele) that is incapable of initiating a p53-dependent apoptosis but is competent to execute p53-mediated cell-cycle arrest/replicative senescence allowed this question to be addressed directly.19 In this system, dysfunctional telomeres potently induced p53-mediated replicative senescence to suppress spontaneous tumorigenesis, while p53-dependent apoptosis was dispensable for tumor suppression. Dysfunctional telomere-induced senescence was accompanied by robust increase in p53, p21 and SA-β-gal activity in all tissue compartments examined, indicating that a telomere-dependent DDR is activated in vivo. In a corresponding study, telomere-induced senescence also suppressed lymphoma formation in mice possessing dysfunctional telomeres in which the apoptotic function of p53 has been eliminated by overexpression of Bcl2.20 Taken together, these two findings firmly established that cellular senescence initiated by dysfunctional telomeres does exist in vivo and potently suppress tumorigenesis in mice.21 Indeed, recent observations indicate that the DDR is activated at the earliest stages in many human adenomas by diverse genotoxic stress to suppress further tumor progression.22 These results predict that activation of an intact DDR pathway by dysfunctional telomeres would promote p53-dependent senescence, suppressing further tumor progression. An intriguing question is whether initiation of cellular senescence results in permanent growth arrest of premalignant lesions in vivo. Is it possible for would-be cancer cells to eventually escape the growth constraints of cellular senescence? Senescent mouse cells in which p53 is ectopically eliminated can indeed resume growth.23, 24 However, cellular senescence mediated by p16 in human cells cannot be reversed.23 Since senescent cells can also secrete pro-inflammatory factors that promote malignant transformation of neighboring cells,25 it is possible that tumor suppression mediated by cellular senescence is actually a double-edged sword: while senescence acts to potently suppress tumorigenesis, the accumulation of senescent cells in vivo may facilitate the development of cancer as we age.
Role of telomeres and telomerase in tumor initiation and progression
The rare cells that stochastically lose p53 function could escape the senescence checkpoint and continue to shorten their telomeres, resulting in entry into a phase of rampant chromosomal instability termed crisis, characterized by chromosomal fusions and NRTs.26, 27 Depending on how fused chromosomes are resolved, loss of heterozygosity (LOH) of tumor suppressors and/or amplification of oncogenes could lead to a pro-cancer genotype. Virally transformed human cells that eliminate p53 and/or pRB function escape crisis at extremely low frequencies,28 while those expressing hTERT are readily immortalized.29, 30 Cell-culture transformation assays showed that p53 null mouse cells with critically shortened telomeres exhibit increased susceptibility to transformation by Myc and Ras oncogenes.31 Similar findings were observed in vivo, in which telomerase-null mice with dysfunctional telomeres and loss of p53 resulted in the selection of cells with a pro-oncogenic genome and early onset of cancer.31 Moreover, while 80–90% of human tumors possess telomerase activity, the remainder maintains telomeres via a recombination-mediated process termed ALT (for alternative lengthening of telomeres) that is telomerase-independent.32, 33, 34 Together, these observations reinforce the importance of an intact p53 pathway in tumor prevention, and support the view that crisis provides a potent barrier to tumor development and, by extension, that telomere maintenance is an essential aspect of full malignant progression.
In human aging populations, cancer deaths are primarily due to carcinomas of the breast, lung and colon, which arise from the epithelial compartment. Loss of p53 function characterizes most human carcinomas, where p53 mutations are found in approximately 50% of human breast adenocarcinomas and 40–60% of colorectal adenocarcinomas.6, 35, 36 However, carcinomas are rarely observed in mice, which normally succumb to lymphomas and sarcomas. This species-specific difference in the tumor spectrum may be due to differences in the length of telomeres and how telomerase levels are regulated, since the long telomeres in mice and somatic telomerase expression would normally prevent the generation of dysfunctional telomeres. Intriguingly, a few carcinomas were observed in telomerase-null mice with dysfunctional telomeres indicating a possible role for telomere dysfunction in promoting tumorigenesis in epithelial compartments. However, the rapid death of telomerase-null, p53−/− mice from lymphomas and sarcomas masked the impact of telomere dysfunction and the ensuing genetic instability in renewing epithelial compartments.
To uncover a potential link between telomere dysfunction and carcinoma development in vivo, telomerase-null, p53+/− mice with a much longer tumor latency were used to further evaluate the impact of age-related telomere shortening on the tumor spectrum of mice. Strikingly, carcinomas emerged as the largest class of clinically apparent tumors, and their cytogenetic profiles resemble those of human carcinomas. Breast, colon and squamous cell carcinomas isolated from telomerase-null, p53+/− mice with telomere dysfunction revealed the presence of multiple chromosomal structural aberrations, including NRTs and amplifications of the distal portion of chromosome 6.37, 38 This region of the mouse chromosome contains the k-ras2 proto-oncogene, which was amplified in all tumors examined and most likely confers growth advantage properties to these lesions.38 Recent high-resolution genomic studies of lymphomas derived from p53/ATM/telomerase triple null mice with dysfunctional telomeres revealed that the emergent murine T-cell lymphomas possess complex genomic alterations similar to those found in human lymphomas and carcinomas, including deletion/mutation of PTEN and FBX7 loci.39 This result suggest that telomere dysfunction engender genomic rearrangements that likely impinge upon cancer-relevant pathways that may allow for the stepwise accumulation of genetic changes in favor of tumor progression, allowing selection by would-be cancer cells of more aggressive traits, such as the ability to elicit an angiogenic response, metastasize, and ultimately survive the actions of chemotherapeutic drugs. In human breast cancers, telomere shortening occurs early during breast tumorigenesis, and genomic instability fueled by dysfunctional telomeres is associated with the transition from benign ductal hyperplasia to malignant ductal carcinoma in situ (Figure 1c).40, 41 Telomerase is reactivated in the majority of advanced human carcinomas,41, 42 suggesting that telomerase reactivation is a critically important step for initiated lesions to progress to frank malignancies since it removes the short telomere barriers inhibitory for tumor progression. Together, these findings demonstrate that telomere dysfunction promotes chromosomal fusions that facilitate the development of carcinomas in the setting of p53 deficiency, and may play a key role in driving genomic instability observed in human carcinomas lacking p53.
Therapeutic opportunities in the telomere field
The upregulation of telomerase in most human cancers and its strict requirement for tumor proliferation makes telomerase an attractive target for cancer therapeutics. Two emerging antitelomerase therapies that have shown promise in animal and clinical trials are highlighted here. Other antitelomerase therapies are discussed in a recent review.43
Anti-hTERT Immunotherapy
The nearly universal expression of hTERT in human cancers, its important role in promoting tumor growth and its restricted expression in normal tissues makes hTERT an attractive antitumor target.44 Although hTERT is expressed at low levels in normal cells, it is able to elicit a cytotoxic T-lymphocyte (CTL)-mediated immune response against tumors expressing high levels of hTERT. Several phase I/II studies have been conducted with hTERT vaccines in patients with advanced lung and pancreatic cancers, and the results are encouraging. Immunotherapies using hTERT peptides GV1001 and HR2822 in combination with granulocyte macrophage-colony stimulating factor (GM-CSF) as an injectable vaccine into patients with non-small cell lung cancer stimulated an immune response in 11 of 24 patients.45 One patient that developed GV1001-specific CTLs exhibited a complete tumor response. Cloned GV1001-specific CD4+ T-cells proliferated specifically against GV1001-pulsed antigen presenting cells and was able to kill them in vitro, demonstrating that the responding T cells are able to efficiently recognize hTERT epitopes. In the trial involving non-resectable pancreatic cancer, patients received intradermal injections of one of three doses of GV1001 in combination with GM-CSF for 10 weeks, followed by monthly booster injections.46 Immune response was observed in 24 of 38 patients, with patients receiving the intermediate dose experiencing the greatest response. This group of patients also experienced a significantly increased survival (median survival 8.6 months, P=0.006). In addition, immune responders lived significantly longer than non-responders (7.2 vs 2.9 months, P=0.001). Importantly, both of these studies demonstrate that hTERT peptides are well tolerated and did not exhibit bone marrow toxicity or other major side effects.
GRN163L Oligonucleotide-Based Therapy
GRN163L is a lipidated 13-mer modified oligonucleotide complementary to the template region of hTERC and is a potent and specific telomerase antagonist.47 Administration of GRN163L to rodent xenograph models bearing either human lung cancer cells,48 human hepatoma cell lines,49 and U-251 human glioblastoma cells50 all resulted in inhibition of tumor progression. Progressive telomere shortening was observed in GRN163L treated hepatoma and lung tumor cell lines, consistent with the oligonucleotide's antitelomerase effects. GRN163L was recently approved to enter phase I/II clinical trials in chronic lymphocytic leukemia administered on a weekly IV dosing schedule.
Summary
Telomeres and telomerase have been the subject of intense research focus in the past decade because the proper maintenance of telomeres is thought to be important to prevent the early onset of cancer. These studies have shed light on the structure of telomeres and how telomeres are shielded from activating the DDR pathway. In the setting of telomere dysfunction, the status of p53 appears critical for tumor suppression. While it is well known that activation of a p53-dependent apoptotic program is tumor suppressive, it now appears that activation of a p53-dependent cellular senescence program by dysfunctional telomeres also potently suppress tumorigenesis in vivo. Future challenges include understanding why certain tissues engage an apoptotic response while others seem to be culled by activation of cellular senescence, and whether senescent cancer cells are permanently growth arrested in vivo, or is escape into malignancy possible. On the therapeutic front, antitelomerase therapies might emerge as effective treatments for cancer with minimal side effects to rapidly dividing normal cellular compartments.
References
Henderson BE, Bernstein L . Endogenous and exogenous hormonal factors. In: Harris JR, Lippman ME, Morrow M, Hellman S (eds). Diseases of the Breast. Lippincott-Raven: Philadelphia, 1996.
Armitage P, Doll R . The age distribution of cancer and a multi-stage theory of carcinogensis. Br J Cancer 1954;8:1–12.
Lengauer C, Kinzler KW, Vogelstein B . Genetic instabilities in human cancers. Nature 1998;396:643–649.
Loeb LA . Mutator phenotype may be required for multistage carcinogenesis. Cancer Res 1991;51:3075–3079.
Cahill DP, Lengauer C, Yu J, et al. Mutations of mitotic checkpoint genes in human cancers. Nature 1998;392:300–303.
Hollstein M, Sidransky D, Vogelstein B, et al. p53 mutations in human cancers. Science 1991;253:49–53.
Keohavong P, Gao W, Madhy H, et al. Analysis of p53 mutations in cells taken from paraffin-embedded tissue sections of ductal carcinoma in situ and atypical ductal hyperplasia of the breast. Cancer Lett 2004;212:121–130.
de Lange T . Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev 2005;19:2100–2110.
Harley CB, Futcher AB, Greider CW . Telomeres shorten during ageing of human fibroblasts. Nature 1990;345:458–460.
Allsopp RC, Vaziri H, Patterson C, et al. Telomere length predicts replicative capacity of human fibroblasts. Proc Natl Acad Sci USA 1992;89:10114–11018.
Hayflick L . The limited in vitro lifetime of human diploid cell strains. Exp Cell Res 1965;37:614–636.
Dimri GP, Lee X, Basile G, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA 1995;92:9363–9367.
Wright WE, Shay JW . Time, telomeres and tumours: is cellular senescence more than an anticancer mechanisms? Trends Cell Biol 1995;5:293–297.
Campisi J . Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell 2005;120:513–522.
Schmitt CA . Senescence, apoptosis and therapy–cutting the lifelines of cancer. Nat Rev Cancer 2003;3:286–295.
Blasco MA, Lee HW, Hande MP, et al. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA [see comments]. Cell 1997;91:25–34.
Greenberg RA, Chin L, Femino A, et al. Short dysfunctional telomeres impair tumorigenesis in the INK4a(delta2/3) cancer-prone mouse. Cell 1999;97:515–525.
Gonzalez-Suarez E, Samper E, Flores I, et al. Telomerase-deficient mice with short telomeres are resistant to skin tumorigenesis. Nat Gen 2006;26:114–117.
Cosme-Blanco W, Shen S, Lazar A, et al. Telomere dysfunction suppresses spontaneous tumorigenesis in vivo by activating p53-mediated cellular senescence. EMBO Reports 2007;8:497–503.
Feldser DM, Greider CW . Short telomeres limit tumor progression in vivo by inducing senescence. Cancer Cell 2007;11:461–469.
Sedivy JM . Telomeres limit cancer growth by inducing senescence: long-sought in vivo evidence obtained. Cancer Cell 2007;11:389–391.
Bartkova J, Horejsi Z, Koed K, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005;434:864–870.
Beausejour CM, Krtolica A, Gamlimi F, et al. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J 2003;22:4212–4222.
Sage J, Miller AL, Perez-Mancera PA, et al. Acute mutation of retinoblastoma gene function is sufficient for cell cycle re-entry. Nature 2003;424:223–228.
Krtolica A, Parrinello S, Lockett S, et al. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci USA 2001;98:12072–12077.
Counter CM, Avilion AA, LeFeuvre CE, et al. Telomere shortening associated with chromosome instability is arrested in immortal cells which express telomerase activity. EMBO J 1992;11:1921–1929.
Chang S, Khoo C, Naylor M, et al. Telomere-based crisis: functional differences between telomerase activation and ALT in tumor progression. Genes and Development 2003;17:88–100.
Shay JW, Van Der Haegen BA, Ying Y, et al. The frequency of immortalization of human fibroblasts and mammary epithelial cells transfected with SV40 large T-antigen. Exp Cell Res 1993;209:45–52.
Bodnar AG, Ouellette M, Frolkis M, et al. Extension of life-span by introduction of telomerase into normal human cells. Science 1998;279:349–352.
Vaziri H, Benchimol S . Reconstitution of telomerase activity in normal human cells leads to elongation of telomeres and extended replicative life span. Curr Biol 1998;8:279–282.
Chin L, Artandi SE, Shen Q, et al. p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell 1999;97:527–538.
Kim NW, Piatyszek MA, Prowse KR, et al. Specific association of human telomerase activity with immortal cells and cancer. Science 1994;266:2011–2015.
Bryan TM, Englezou A, Dalla-Pozza L, et al. Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines. Nat Med 1997;3:1271–1274.
Shay JW, Bacchetti S . A survey of telomerase activity in human cancer. Eur J Cancer 1997;33:787–791.
Greenblatt MS, Bennett WP, Hollstein M, et al. Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis. Cancer Res 1994;54:4855–4878.
Veloso M, Wrba F, Kaserer K, et al. p53 gene status and expression of p53, mdm2, and p21Waf1/Cip1 proteins in colorectal cancer. Virchows Arch 2000;437:241–247.
Artandi S, Chang S, Lee S, et al. Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature 2000;406:641–645.
O'Hagan RC, Chang S, Maser RS, et al. Telomere dysfunction provokes regional amplification and deletion in cancer genomes. Cancer Cell 2002;2:149–155.
Maser RS, Choudhury B, Campbell PJ, et al. Chromosomally unstable mouse tumours have genomic alterations similar to diverse human cancers. Nature 2007;447:966–971.
Chin K, de Solorzano CO, Knowles A, et al. In situ analyses of genome instability in breast cancer. Nat Genet 2004;36:984–988.
Meeker AK, Argani P . Telomere shortening occurs early during breast tumorigenesis: a cause of chromosome destabilization underlying malignant transformation? J Mammary Gland Biol Neoplasia 2004;9:285–296.
Tsao JL, Zhao Y, Lukas J, et al. Telomerase activity in normal and neoplastic breast. Clin Cancer Res 1997;3:627–631.
Shay JW, Wright WE . Telomerase therapeutics for cancer: challenges and new directions. Nat Rev Drug Discov 2006;5:577–584.
Carpenter EL, Vonderheide RH . Telomerase-based immunotherapy of cancer. Expert Opin Biol Ther 2006;6:1031–1039.
Brunsvig PF, Aamdal S, Gjertsen MK, et al. Telomerase peptide vaccination: a phase I/II study in patients with non-small cell lung cancer. Cancer Immunol Immunotherapy 2006;55:1553–1564.
Bernhardt SL, Gjertsen MK, Trachsel S, et al. Telomerase peptide vaccination of patients with non-resectable pancreatic cancer: a dose escalating phase I/II study. Br J Cancer 2006;95:1474–1482.
Dikmen ZG, Gellert GC, Jackson S, et al. In vivo inhibition of lung cancer by GRN163L: a novel human telomerase inhibitor. Cancer Res 2005;65:7866–7873.
Jackson SR, Zhu CH, Paulson V, et al. Antiadhesive effects of GRN163L—an Oligonucleotide N3′ → P5′ Thio-Phosphoramidate targeting Telomerase. Cancer Res 2007;67:1121–1129.
Djojosubroto MW, Chin AC, Go N, et al. Telomerase antagonists GRN163 and GRN163L inhibit tumor growth and increase chemosensitivity of human hepatoma. Hepatology 2006;42:1127–1136.
Ozawa T, Gryaznov SM, Hu LJ, et al. Antitumor effects of specific telomerase inhibitor GRN163 in human glioblastoma xenografts. Neuro-oncol 2004;6:218–226.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Deng, Y., Chang, S. Role of telomeres and telomerase in genomic instability, senescence and cancer. Lab Invest 87, 1071–1076 (2007). https://doi.org/10.1038/labinvest.3700673
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.3700673
Keywords
This article is cited by
-
The dynamics of telomere length in primary and metastatic colorectal cancer lesions
Scientific Reports (2023)
-
Exploiting senescence for the treatment of cancer
Nature Reviews Cancer (2022)
-
Telomerase gene therapy: a remission toward cancer
Medical Oncology (2022)
-
Cancer gene therapy by NF-κB-activated cancer cell-specific expression of CRISPR/Cas9 targeting telomeres
Gene Therapy (2020)
-
Bioinformatic framework for analysis of transcription factor changes as the molecular link between replicative cellular senescence signaling pathways and carcinogenesis
Biogerontology (2020)
