
Abstract
The hepatoprotective effect of interleukin-6 (IL-6)/signal transducer and activator of transcription 3 (STAT3) has been well documented. However, reports on the role of IL-6/STAT3 in liver regeneration are conflicting probably due to the fact that the model of Stat3 knockout mice were complicated with obesity and fatty liver, which may cause some secondary effects on liver regeneration. To study the direct role of STAT3 and to circumvent the problems of obesity and fatty liver in liver regeneration, we generated conditional STAT3 knockout in the liver (L-Stat3−/−) using a transthyretin-driven Cre-lox method. The L-Stat3−/− mice were born with the expected Mendelian frequency and showed no obesity or other obvious phenotype. After partial hepatectomy, mortality in the L-Stat3−/− mice was significantly higher than the littermate Stat3f/+ controls in the early time points (<24 h). Hepatocyte DNA synthesis in the survived L-Stat3−/− mice slightly decreased as compared with Stat3f/+ mice at 40 h after partial hepatectomy, whereas similar hepatocyte DNA synthesis was found at other time points and liver mass could be completely recovered in the L-Stat3−/− mice. In another model of liver regeneration induced by subcutaneous injection of carbon tetrachloride (CCl4), hepatocyte DNA synthesis in the CCl4-treated L-Stat3−/− mice also decreased as compared with Stat3f/+ mice at 40 h after injection but not at other time points. In addition, infiltration of neutrophils and monocyte increased in the liver of CCl4-treated L-Stat3−/− mice compared to wild-type mice. In conclusion, STAT3 is required for survival in the acute stage after 70% hepatectomy and plays a role in inflammatory reaction after hepatocyte necrosis. However, the hepatocytic STAT3 may have limited role in liver mass recovery although DNA synthesis may be impaired.
Similar content being viewed by others


Main
The liver is the only organ that has the ability to regenerate after massive loss of tissue mass induced by partial hepatectomy (PH) or toxic injury. The regeneration is achieved by the remaining hepatocytes that are normally quiescent in the healthy liver, but have the capacity to proliferate after liver damage for restoring the liver mass within 7–10 days.1, 2 In the past years manifold studies implicated important roles of different cytokines and growth factors for hepatocyte proliferation, such as insulin, glucagon, epidermal growth factor (EGF), hepatocyte growth factor, transforming growth factor-β, fibroblast growth factor, interleukin-6 (IL-6) and tumor necrosis factor-α.1, 2 Studies using knockout mice lacking either type I tumor necrosis factor receptor I or IL-6 showed a defect in liver regeneration, underlining the importance of these cytokines in vivo.3, 4 However, increasing evidence suggests that IL-6 plays more important role in protection against liver injury, and only a minor role in liver regeneration.5, 6
Several transcription factors are activated shortly after PH or toxic liver injury, including NF-κB, signal transducer and activator of transcription 3 (STAT3), AP-1 and C/EBP.4, 7, 8, 9, 10 STAT3 belongs to the STAT family of transcription factors which consists of at least seven members in mammals.11 STAT3, initially named acute-phase response factor, was shown to be activated by all members of the IL-6 family, including IL-6, IL-11, oncostatin M (OSM), leukemia inhibitory factor, cardiotropin 1 and ciliary neutrophic factor. The targeted disruption of Stat3 leads to early embryonic lethality, indicating an essential function of Stat3 in embryonic cell growth.12 In the liver, activation of STAT3 by IL-6, its related cytokines, and IL-22, plays an important role in acute-phase response and hepatoprotection.13, 14, 15, 16, 17, 18, 19 Early data showed that STAT3 increased after PH but it was not just associated with the regenerative phase.20 Later Li et al21 reported that hepatocyte DNA synthesis at 40 h posthepatectomy in Alb(+) Stat3(fl/fl) livers was reduced to approximately one-third of the control. Recently, in the study using the Alb(+) Stat3(fl/fl) mice, Haga et al22 concluded that PI3-K/Akt-mediated responsive hepatocellular hypertrophy may be essential for liver regeneration following hepatectomy and sufficiently compensated liver regeneration in STAT3-deficient liver. These elegant studies explored the role of STAT3 in liver regeneration and suggested that hepatocellular hypertrophy may be essential for liver regeneration following hepatectomy in STAT3-deficient mice.22 Because hypertrophy or increase in cell size is largely a metabolic event, rather than a regenerative process, which involves cell division, the role of STAT3 in liver regeneration need to be further defined especially when the animal model was complicated with obesity and fatty liver,22, 23 which might cause some secondary effects on liver regeneration.24, 25 In this study we describe the generation of a strain of lean mice with liver-STAT3 deficiency and show direct roles of STAT3 in liver injury and regeneration.
Materials and methods
Animals
All mice were maintained according to the criteria outlined in the guide for the care and use of laboratory animals published by the National Institutes of Health. Experiments using mice were approved by Yale Animal Care and Use Committee and Institutional Animal Care and Use Committee of Indiana University Medical School.
Generation of Transgenic Mice Expressing Cre Recombinase in Liver
Vector TTR1ExV3, which contains the upstream transcriptional regulation region, the first exon and intron, and a partial second exon of the transthyretin (Ttr) gene,26 was digested with StuI (Figure 1a). A fragment containing the opening reading frame (ORF) of the Cre gene was recovered from the mammalian expression vector pBS185 (GIBCO BRL) by digestion with XhoI and MluI and inserted into the StuI site of TTR1ExV3 to get the final construct pTtr-Cre. Plasmid pTtr-Cre was digested with HindIII to generate a transgene fragment, which was isolated by agarose gel electrophoresis. The purified transgene fragment was injected into fertilized eggs as described previously.27 Six Ttr-Cre transgenic founders were identified by PCR and crossed with mice carrying a DNA polymerase β-allele containing two loxP sites to examine the efficiency and tissue specificity of Cre-dependent deletion (Figure 1b). Strain Ttr10-3 shows Cre recombinase activity specifically in liver and digestive tissues (Figure 1c).

Generation of transgenic mice expressing Cre recombinase in the liver. (a) Construction of Ttr-Cre transgene expression vector: the Ttr-Cre transgene expression vector contains a Ttr minigene consisting of 3.0 kb upstream regulatory region, the transcription initiation site, the first exon and intron and a part of the second exon followed by an SV40 polyadenylation sequence. A 1.1-kb fragment containing the Cre cDNA was inserted into the StuI site of exon 2. DNA containing the transgene was injected into oocytes from C57BL/6 mice to generate transgenic mice. TATA: TATA-box; ATG: start codon; polyA: SV40 polyadenylation sequence. (b) Genomic map of mutant and wild-type polβ alleles used for Cre recombinase activity detection: deletion of the floxed DNA segment results in removal of the promoter and first exon of polβ. The produce of this deletion is detected as a 3.5-kb band after BamHI digestion. polβ+: wild-type polβ allele; polβF: floxed allele; polβ–: deleted allele; P: promoter; E1: exon1, E2: exon2, black arrow: loxP site. (c) Strain Ttr10-3 shows Cre recombinase activity specifically in liver and digestive tissues: Ttr-Cre transgenic mice (Ttr10-3) were crossed with mice carrying a DNA polβ in a polβF configuration. Different tissues of genomic DNA from adult offspring were digested with BamHI and subjected to Southern blot analyses using polβ-specific probe.
Generation of Stat3flox Mice
Genomic clones containing the SH2 domain of the murine Stat3 gene were isolated from a 1Fix II murine 129/Sv genomic library by screening the library using the StuI–ScaI fragment of the murine Stat3 cDNA as a probe. Six-phage clones containing SH2 domain sequences of the murine Stat3 gene were identified.
Procedures for the construction are shown in Figure 2a. The targeting vector was constructed using a 3.8 Kb SmaI fragment, a 2.7 Kb NotI–SmaI fragment and a 10 Kb NotI–SpeI fragment of clone 1, which were inserted into pBluescript KS to generate pBS-1-A, pBS-1-B and pBS-1-C, respectively. DNA sequencing and PCR analyses showed that pBS-1-A contained exons 20 (3′ end), 21 and 22, and that pBS-1-B contained exons 15, 16, 17, 18, 19 and 20 (5′ end). The SH2 domain was found to be coded by exons 19 and 20. A loxP site coupled with a HindIII restriction site was introduced into intron 20 of pBS-1-A by insertional mutagenesis (Construct I). Because introns 18 and 19 are too short to introduce another loxP site without regard to interference of splicing, we produced a BglII restriction site in intron 17 of pBS-1-B by site-directed mutagenesis and inserted a neomycin-resistance gene cassette (neor) flanked by two loxP sites into the BglII site (Construct II). An HSV-thymidine kinase gene cassette (HSV-tk) was inserted into the SpeI site of pBS-1-C to get Construct III. A SmaI fragment was excised from Construct I and inserted into the SmaI site of Construct II to get Construct IV. A Kpn2I–SalI fragment was excised from Construct III and inserted into the Kpn2I–SalI site of Construct IV to get the final construct, pNT-Stat3.

Generation of Stat3flox mice. (a) Schematic strategy for generation of targeting vector and conditional Stat3 alleles: the targeting vector was constructed by using genomic clones from a 1FixII murine 129/Sv genomic library. A loxP site coupled with a HindIII restriction site was introduced into intron 20 by insertional mutagenesis (construct I). The neomycin-resistance gene, flanked by two loxP sites, was inserted into intron 17 (construct II). An HSV-thymidine kinase gene cassette (HSV-tk) was inserted into the SpeI site of pBS-1-C to get Construct III. A Kpn2I–SalI fragment was excised from Construct III and inserted into the Kpn2I–SalI site of Construct IV to get the final construct. For further details see Materials and methods. (b) Generation of Stat3 flox mice: to delete the floxed neoR gene, heterozygous Stat3neo-flox mice were bred to deleter strain Ttr10-1. Genomic DNA from tail was digested with EcoRI and HindIII, separated by agarose gel electrophores, and then hybridized with radiolabeled probe A. The mice that lack neoR and remain 2 loxp sites flanking exons 18–20 were identified by a 4.8 kb band in the Southern blot. (c) Generation of tissue-specific Stat3 knockout mice: homozygous Stat3flox/flox mice were bred to strain Ttr10-3. Genomic DNA from Cre+; Stat3flox/+ and Stat3flox/+ mice without Cre recombinase was obtained from tail and liver, digested with BglII and analyzed by Southern blot using probe B. Successful deletion in the Stat3 gene was detected in the liver of Cre+; Stat3flox/+ mice as indicated by the appearance of a 1.8 kb band (Stat3D).
The W9.5 embryonic stem cell line was cultured as described.28 Embryonic stem cells were electroporated with 25 μg of SalI-linearized pNT-Stat3 per 1 × 107 cells and grown under double selection as described.28 To identify ES cells carrying a homologous recombination of the targeting fragment, genomic DNA was isolated and digested with HindIII and analyzed by Southern blot with probe A. Three clones with 4.8 Kb bands were found and micro-injected into blastocysts of C57BL/6 mice to generate chimeric mice.
Genotype Analysis
Three different primer pairs were routinely used to detect the Stat3flox and Stat3D alleles and the inserted Cre gene by PCR analysis. The Stat3flox allele was detected using primers 1 (5′-ATTGGAACCTGGGACCAAGTGG) and 2 (5′-ACATGTACTTACAGGGTGTGTGC), which amplified a 520-bp fragment, while Stat3 wild-type allele gave 480-bp fragment. The Stat3D allele was detected as a 480-bp fragment with primers 1 and 3 (5′-GCTGGCTCATAGGCAAAAACAC). The Ttr-Cre transgene was detected using the primers 5′-CCAGCTAAACATGCTTCATCGTCGTC and 5′-ATTCTCCCACCGTCAGTACGTGAG, which amplified a 300-bp fragment.
Isolation of Hepatocytes
Hepatocytes were isolated by in situ perfusion as described previously29 with minor modifications. Briefly, livers were perfused with EGTA (0.5 mM) and collagenase (0.15 g/l) via the inferior cava. Following perfusion, the dissociated liver was minced and suspended in HBSS solution. Cells were centrifuged twice at 500 g and hepatocytes were separated by centrifugation through a Percoll layer. Cells were counted and plated onto ∅10 cm dishes with 2 × 106 cells.
PH and CCl4 Injection
Seventy percent hepatectomy (PH) was performed on L-Stat3−/− (Cre+; Stat3flox/flox) and Stat3f/+ mice (Cre−; Stat3flox/+ and Stat3flox/D) under Ketamine (100 mg/kg) and Xylazine (10 mg/kg) anesthesia by ligating and removing the left lateral and median two lobes of the liver, including the gallbladder. Sham-operated mice underwent mid-ventral laparotomy without excision of the liver. The remnant livers were harvested between 1 h and 1 month after PH according to designed experiments. Liver tissue was immediately frozen in dry ice or placed in 4% paraformaldehyde in PBS for paraffin embedding.
For CCl4-induced liver injury, 3 ml/kg of 50% CCl4 (Sigma Chemical Co., St Louis, MO, USA) was injected with sterile olive oil subcutaneously. Free access to food and water was allowed after injection.
BrdU Labeling and Histological Staining
BrdU (Sigma Chemical Co.) was injected into the peritoneum at a concentration of 50 μg/g body weight. The animals were sacrificed 2 h after injection. The liver was immersed in Tissue-Tek® OCT compound (Sakura Finetek USA, Torrance, CA, USA) and snap frozen in acetone cooled with dry ice. The liver was sectioned with a cryotome and fixed in acetone for 15 min. Labeled cells were localized with an anti-BrdU monoclonal antibody (Sigma), using routine indirect immunoperoxidase staining. The number of hepatocytes undergoing DNA synthesis was counted by randomly counting the number of BrdU-labeled hepatocytes per 1000 hepatocytes. Tissue sections from 4 to 6 different L-Stat3−/− and heterozygous Stat3f/+ mice were evaluated at 24, 40, 48 and 72 h after PH.
For hematoxylin and eosin (HE) staining, the sections were prepared using the method above and sent to Yale Pathological Services for the staining.
Tissue Extraction and Immunoblotting
Tissues were homogenized in whole-cell extraction buffer (400 mM KCl, 10 mM NaHPO4, 1 mM EDTA, 1 mM dithiothreitol (DTT), 10% (w/v) glycerol, 1 μg/ml aprotinin, 1 μg/ml leupeptin, 1 μg/ml pepstatin, 1 mM phenylmethylsulfonyl fluoride (PMSF), 5 mM NaF and 1 mM Na3VO4). After centrifugation at 14 000 r.p.m., the supernatant was collected, and protein concentration was quantified (BCA kit, PIERCE). Ten-micrograms of protein were separated by SDS-polyacrylamide gel electrophoresis and then transferred to Immuno-Blot™ polyvinylidene difluoride membrane (Bio-Rad). After blocking with 5% nonfat milk in washing buffer (10 mM Tris–HCl, 150 mM NaCl, 1 mM EDTA and 0.1% (w/v) Triton X-100, pH 8.0), the membranes were soaked in the indicated antibodies, followed by soaking in anti-rabbit or anti-mouse IgG coupled with horseradish peroxidase and visualized by using SuperSignal® Chemiluminescent Substrate (PIERCE).
Preparation of Nuclear Extracts from Whole Cells and Tissue
Nuclear extract preparation was performed as described previously.20 Briefly, hepatocytes or liver tissue were washed with PBS and homogenized in buffer A (0.32 M sucrose, 10 mM HEPES (pH 7.9), 25 mM KCl, 0.2 mM spermine, 0.5 mM spermidine, 1 mM EDTA, 1 mM EGTA, 1 mM PMSF, 1 mM DTT, 1 mM Na3VO4, 1 μg/ml leupeptin, 2 μg/ml aprotinin and 1 μg/ml pepstatin). The homogenates were mixed with buffer B (buffer A plus 2 M sucrose) and ultra-centrifuged at 20 000 r.p.m. for 50 min at 4°C. Pellets were resuspended in 50 μl nuclear extraction buffer A (10 mM HEPES (pH 7.9), 25 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 20% glycerol, 0.5 mM PMSF, 1 mM DTT, 1 mM Na3VO4, 2 μg/ml leupeptin, 2 μg/ml aprotinin and 2 μg/ml pepstatin). Fifty microliters of lysate buffer (extraction buffer A plus 815 mM KCl) was added and the samples were incubated on ice for 30 min. After centrifugation at 1400 r.p.m., the supernatants were collected and protein concentrations were quantified.
Electrophoretic Mobility Shift Assay
Tissue or cell extracts were obtained as described above. Ten microgram of whole cell or 1 μg of nuclear extract was mixed with 5′-end 32P-labeled m67-SIE probe (0.1 ng) in buffer containing 13 mM HEPES (pH 7.9), 185 mM NaCl, 0.15 mM EDTA, 8% glycerol, 1 μg of single-stranded DNA and 1 μg poly (dI-dC), and incubated for 20 min at room temperature. DNA–protein complexes were separated on 5% nondenaturing polyacrylamide gels in 0.5 × Tris-Borate-EDTA buffer and detected by autoradiography.
Statistical Analysis
Data are presented as mean±s.d. The difference was analyzed by a paired Student's t-test. Data in a time cause experiment were tested by ANOVA. Probability less than 0.05 was considered to be significant.
Results
Generation of Transgenic Mice Expressing Cre Recombinase Specifically in Liver
To elicit targeted expression of Cre in liver, we used transcriptional regulatory sequences from the Ttr promoter. The Ttr gene encodes for the liver-specific TTR, a serum thyroid or vitamin A carrier produced in hepatocytes.27 The Ttr-Cre transgene was generated by inserting the Cre coding sequence into the second exon of a Ttr expression vector previously used to drive transgene expression in the liver (Figure 1a).27 To examine the efficiency and tissue specificity of Cre-dependent deletion, Ttr-Cre transgenic mice were crossed with mice carrying a DNA polymerase β-gene (polβ) in a floxed (polβF) germline configuration. DNA from adult offspring was subjected to Southern blot analyses using polβ-specific probe (Figure 1b). Deletion of the floxed DNA segment resulted in removal of the promoter and first exon of polβ, leading to the generation of a 3.5 kb band. As expected, one line (Ttr19-1) showed a liver-specific recombination of the loxP sites with a deletion efficiency of 50% (data not shown). Such mosaic expression in adult liver was also observed in p53 dominant-negative transgenic mice with the Ttr promoter.30 Another line, Ttr10-3, directed deletion of the loxP sites in pancreas, gut and gallbladder in addition to liver, with efficiencies of 100, 50, 50 and 95%, respectively (Figure 1c). TTR has recently been shown to be expressed in pancreas31 and fetal gut.32, 33
Generation of Stat3 Conditional Knockout Mice
To overcome the embryonic lethality of Stat3 gene disruption, we generated tissue-specific Stat3 knockout mice using the Cre-loxP recombinase system. For this purpose we created a conditional Stat3 gene allele (Stat3neo-flox) in ES cells containing three loxP sequences by gene targeting (Figure 2). Two loxP sequences flank exons 18, 19 and 20 of the murine Stat3 allele, whereas the third loxP site is located upstream of the neomycin-resistance gene (neoR), which is integrated into intron 17 (Figure 2a, Stat3neo-flox). Since exons 19 and 20 harbor the coding region for the SH2 domain, Cre recombinase activity produces a mutant STAT3 protein lacking this essential domain for STAT3 function (Figure 2a, Stat3D). ES cell clones showing a homologous recombination of the targeting construct were identified by Southern blot analysis (data not shown) and microinjected into blastocytes of C57BL/6 mice to generate chimeric mice. To delete the neoR gene, heterozygous Stat3neo-flox mice were bred to deleter strain Ttr10-1, which shows a weak Cre recombinase expression in almost all tissues including germline cells. Partial Cre-mediated recombination leads to the conversion of the Stat3neo-flox allele to a second conditional allele (Stat3flox, see Figure 2a) that lacks neoR but remains two loxP sites flanking exons 18–20. Three mice showing a mosaic pattern of the Stat3flox allele were obtained by PCR and further confirmed by Southern blot (Figure 2b, representative blot). One of them showed a germline transmission of Stat3flox after breeding with C57BL/6 mice. To eliminate the Cre gene, heterozygous Stat3flox/+ mice were further bred to C57BL/6 mice. Using this method we were also able to obtain heterozygous Stat3 knockout mice in which the Stat3neo-flox allele was converted into a third allele (Stat3D, Figure 2a) lacking exons 18–20 and the neoR gene.
Generation of Mice Lacking Stat3 Specifically in Liver and Digestive Tissues
In order to obtain a more complete deletion of the Stat3 gene in liver, we decided to use strain Ttr10-3 which shows high Cre recombinase activity in liver and pancreas (Figure 1c). To confirm this, Ttr10-3 strain mice were bred with homozygous Stat3flox/flox mice. Samples of liver tissue and tails were extracted and Southern blot was performed to visualize the different Stat3flox alleles with probe B (Figure 2c). Recombination between the flox sites in the Stat3 gene was only found in liver and not in the tail, indicating a tissue-specific deletion of the Stat3 allele.
We continued this strategy to obtain complete deletion of the Stat3 gene. The Ttr10-3 strain was crossed with heterozygous Stat3D/+ mice to generate Cre+; Stat3D/+ mice. The mice were bred with homozygous Stat3flox/flox mice to generate Cre+; Stat3flox/D mice with liver-specific Stat3 deletion (L-Stat3−/−). Cre+; Stat3flox/D mice were born with the expected Mendelian frequency, demonstrating that the absence of Stat3 in liver did not lead to embryonic lethality in these mice. All mice reached adulthood and showed no reduction in life span for up to one and a half years. The liver weight/body weight ratio was not altered in L-Stat3−/− mice (5.5±0.44%) in comparison to Stat3f/+ mice (4.9±0.25%). Histological examination of liver sections showed no obvious differences in L-Stat3−/− mice in comparison to sections obtained from Stat3f/+ mice (data not shown).
Biochemical Evidence of STAT3 Deletion in the Liver
To examine the level of STAT3 protein in different tissues, we performed Western blot analyses. As shown in Figure 3a, full-length STAT3 expression was undetectable in the liver of L-Stat3−/− mice (Cre+ Stat3flox/D), whereas Stat3+/+ and heterozygous Stat3flox/D mice retained STAT3 protein. To confirm this result, we detected the STAT3-specific mRNA in isolated hepatocytes using RT-PCR. In mice lacking functional STAT3 in liver (Cre+ Stat3flox/D) only truncated mRNA was detected, whereas in hepatocytes from heterozygous Stat3flox/D mice, both wild type and truncated form were observed (Figure 3b). We further analyzed the activation of STAT3 after EGF and OSM treatment, since EGF and OSM stimulate phosphorylation and DNA-binding activity of STAT3 in isolated hepatocytes. Electrophoretic mobility shift assay (EMSA) analysis, using the specific m67-SIE binding site as probe, showed the typical formation of DNA-binding complexes consisting of STAT3 and STAT1 homodimers and STAT1/STAT3 heterodimers in extracts from hepatoctyes of C57BL/6 mice after treatment with EGF or OSM, whereas in STAT3-deficient hepatocytes, EGF and OSM treatment failed to induce STAT3 DNA complexes (Figure 3c). However, activation of STAT1 was not impaired in these mice. All the results provide evidence that we have obtained mice lacking functional STAT3 protein in the liver without obesity (Figure 3d).

Liver-specific deletion of STAT3. (a) Full-length STAT3 protein is not detectable in liver of Cre+; Stat3flox/D mice: indicated tissues from Stat3+/+, Cre+; Stat3flox/D and Stat3flox/D mice were homogenized and the supernatant was separated by 8% SDS-PAGE and immunoblotted with anti-STAT3 (H190, Santa Cruz) antibody. The gene product of Cre+; Stat3flox/D mutant was a partial deletion of Stat3 gene (exons 18, 19 and 20). A major band at 90 kDa representing STAT3 is completely and specifically missing in the liver of Cre+; Stat3flox/D mice. (b) STAT3 mRNA is truncated in hepatocytes carrying Stat3 flox or Stat3 D alleles: RT-PCR using RNA isolated from hepatocytes from Stat3+/+, Cre+; Stat3flox/D and Stat3flox/D mice was performed. After reverse transcription cDNA was amplified by PCR using a primer pair binding to exons 13 and 21, respectively. The shorter PCR product (530 bp) shows the presence of a truncated form of STAT3 mRNA in Stat3flox/D mice indicating the specific deletion of the targeted gene fragment. (c) STAT3 binding activity is abolished in hepatocytes from liver-specific Stat3 KO mice: nuclear extracts from Stat3+/+ and Cre+; Stat3flox/D mice stimulated with or without 100 ng/ml hEGF or OSM were incubated with radiolabeled STAT3 binding site (m67-SIE) and were analyzed by EMSA. No STAT3 DNA binding was found in Cre+; Stat3flox/D mice. (d) Body weight of the Cre+; Stat3flox/D (L-Stat3−/−) and Stat3flox/+ mice. The L-Stat3−/− mice were not obese.
Survival Rates and Liver Regeneration after PH in Cre+ Stat3flox/D (L-Stat3−/−) and Control Mice
The mortality rate after PH was significantly higher in L-Stat3−/− mice (Line 10-3) (35.3%) than in Stat3f/+ mice (5.5%) in the first 12 h after PH (Table 1). Nevertheless, the mortality rate >24 h after hepatectomy was not significantly increased in L-Stat3−/− mice. The other line (19-1) had similar results (not shown). As indicated in Figure 4c the time course of liver mass restoration, measured by liver mass/body weight ratios, in L-Stat3−/− mice was similar to that observed in Stat3f/+ mice. BrdU+ hepatocytes were counted in sections of Stat3f/+ and L-Stat3−/− livers. As indicated in Figure 4a and b there was only a marginal decrease in the amount of hepatocytes undergoing DNA synthesis in L-Stat3−/− mice in comparison to Stat3f/+ mice after PH. Histopathological examination of liver sections at different time points after PH shows no obvious differences between L-Stat3−/− and Stat3f/+ mice (Figure 4d).

Liver regeneration after PH. (a) Immunohistochemical staining of BrdU (red) in liver sections at 40 h after PH. Nuclei were counter stained with DAPI (blue). Original magnification: × 40. (b) Summary of Brdu-positive cells after PH. BrdU-labeled hepatocytes were counted 24, 40, 48 and 72 h after PH. For each group (L-Stat3−/− and Stat3f/+ mice) 4–6 mice were used for each time point. *P<0.05 between L-Stat3−/− (closed bar) and Stat3f/+ mice (open bar). (c) Recovery of liver mass after PH. Liver weights were measured at different time points after 70% PH. For each group (L-Stat3−/− and Stat3f/+ mice) 4–6 mice between 10 and 15 weeks old were used for each time point. (d) Histopathological examination of liver sections after PH. HE staining of liver sections from L-Stat3−/− and Stat3f/+ mice was performed at indicated time points after PH. Original magnification: × 20.
Liver Regeneration after CCl4 Injection
The mortality rate after CCL4 injection was not significantly increased in L-Stat3−/− mice (Table 1). As indicated in Figure 5a and b, the necrotic area in the L-Stat3−/− mice was similar to that observed in Stat3f/+ mice. Similar to those in hepatectomy, BrdU-positive hepatocytes significantly decreased in L-Stat3−/− mice in comparison to Stat3f/+ mice (Figure 5c and d). Further more, the L-Stat3−/− mice exhibited significant inflammation at 40 h after CCl4 injection (Figure 6b, the lower right panel shows quantitation and statistics).

Liver regeneration after CCl4 injection. (a) Liver section. HE stain shows hepatocyte necrosis (brighter red area) in L-Stat3−/− and littermate Stat3f/+ controls 40 h after CCl4 injection. (b) Summary of necrotic area. There was no significant difference between the area of necrosis in L-Stat3−/− (n=6, black bar) and Stat3f/+ mice (n=6, white bar) 40 h after CCl4 injection. (c) Summary of BrdU-positive hepatocytes at 40 h after CCl4 injection. BrdU-labeled hepatocytes were counted 24, 40, 48 and 72 h after CCl4 injection. Four to six mice were used for each of the L-Stat3−/− and Stat3f/+ group at each time point. *P<0.01 between L-Stat3−/− and Stat3f/+ mice. (d) Immunohistochemical staining of BrdU (red) in liver sections at 40 h after CCl4 injection. Nuclei were counter stained with DAPI (blue). Original magnification: × 40.

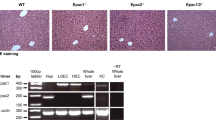
STAT1 activation after hepatectomy and inflammatory infiltration after CCl4 injection. (a) STAT1 and STAT3 DNA binding activity in post hepatectomy liver. Nuclear extracts from liver tissue were obtained at different time points after PH, incubated with radiolabelled m67-SIE oligonucleotides and analyzed by EMSA. Position of STAT3 and STAT1 is indicated by arrows. STAT1 activation is detectable after hepatectomy in KO mice. STAT1: STAT1 homodimer; STAT1/STAT3: STAT1 and STAT3 heterodimer; STAT3: STAT3 homodimer; 3 h sham: liver nuclear extracts from mice 3 h after sham operation. (b) Histopathological examination of liver sections after CCl4 injection. Microphotos: HE staining of liver sections from L-Stat3−/− (n=6) and Stat3f/+ mice (n=6) was performed 40 h after CCl4 injection. For each mouse, 30 sections were made and the microphotos show representative foci of L-Stat3−/− and Stat3f/+ mice. The magnified view shows neutrophil (arrows) and monocyte (arrow heads) infiltration in the liver of L-Stat3−/− mice. Lower right panel: for each mouse, 10 randomized, blinded sections were used to count neutrophils and monocytes according to the distinct morphology under high power magnification (open bars: Stat3f/+, and closed bars: L-Stat3−/− mice). Original magnification: × 40. *P<0.01 between L-Stat3−/− and Stat3f/+.
STAT1 is Activated in Mice Lacking STAT3 in Hepatocytes after PH
To determine the DNA-binding activity of STAT proteins in liver extracts from STAT3-deficient and control mice at different time points after PH, we performed EMSA analysis. As observed in previous studies, STAT3 is strongly activated within 1 h after PH, and the activation persists beyond 8 h (Figure 6a) in the wild-type mice. On the other hand, the induction of STAT1-binding activity at 3 and 8 h after PH is strongly upregulated in liver extracts of STAT3-deficient mice and still detectable after 24 h (Figure 6a).
Discussion
Cre Recombinase Expression and Conditional Knockout of Stat3 in the Liver and Digestive Tissues
To elicit targeted expression of Cre recombinase in the liver, we used transcriptional regulatory sequences from the liver-specific Ttr (encodes for TTR) promoter fused to the Cre ORF to generate transgenic mice (Figure 1a). The Ttr minigene used to drive Cre expression in these experiments has been shown in previous studies to yield nearly the same gene expression pattern as that of the endogenous Ttr gene.27, 34 Namely, in the adult, the transgene and the endogenous gene are expressed in hepatocytes,26, 27, 30 brain choroid plexus epithelium, retina pigment epithelium and pancreatic islets.31 Expression of the Ttr minigene in brain choroid plexus epithelium, however, was shown to require many transgene copies while liver expression is copy number independent.27 The Ttr10-3 Cre line used in this study shows no Cre recombinase activity in the brain (not shown), whereas Cre-dependent recombination could be detected in the liver, pancreas and intestine (Figures 1c and 3a).
We created conditional knockout of Stat3 in the mouse liver (L-Stat3−/−) under the control of Ttr promoter. Unlike liver-specific knockout of Stat3 driven by albumin promoter,23 our L-Stat3−/− mice had neither obesity (Figure 3d) nor fatty liver (Figure 4d). Although the reason for the difference between these two strains of Stat3-deficient mice is unclear, we believe the Ttr-Cre-driven L-Stat3−/− mice are suitable for asking the question about the primary role of STAT3 in liver regeneration because this model makes it possible to circumvent the problems of obesity and fatty liver, which may cause some secondary effects on liver regeneration.24, 25
STAT3 is Required for Survival in the Acute Stage after PH
The death rate in the L-Stat3−/− mice was 6.4-fold of the Stat3f/+ within 24 h after PH (Table 1). We noted that a recent work demonstrates STAT3 contributes to mitogenic activity of hepatocyte after PH.21 Unfortunately, animal mortality in that work is unknown. In the present study, the increased mortality in the L-Stat3−/− mice could not be due to surgical failure because the rate in Stat3f/+ mice was 5.5%, indicating that the operation per se was acceptable. It is known that damages to the liver, including trauma and infection, evoke a rise in the so-called acute-phase proteins in plasma, which are essential for survival after liver injury.35 This acute-phase response is impaired in both IL-64 and STAT315 deficient animals. It is most likely that the L-Stat3−/− mice failed to survive after the PH because there was a disturbance in the acute-phase response, which is fundamentally dependant on STAT3.15
Limited Role of STAT3 in Restoration of Liver Mass and DNA Synthesis after PH and CCl4 Injection
To our surprise, the survived mice lacking STAT3 in hepatocytes could restore the liver mass after PH (Figure 4c) although DNA synthesis in L-Stat3−/− mice was slightly decreased (20.6%) at 40 h after PH, as measured by BrdU incorporation (Figure 4a and b). The decrease in DNA synthesis in the L-Stat3−/− mice was not as dramatic as that in the mice deficient for IL-6, which show over four-fold reduction in Brdu incorporation,4 implicating that hepatocyte proliferation does not rely exclusively on STAT3 and there might be compensatory mechanisms for hepatocyte STAT3. We also examined cell death and liver regeneration in the L-Stat3−/− mice after CCL4 injection. CCl4 did not induce a more severe necrosis in the L-Stat3−/− mice (Figure 5a and b), probably due to the nonspecific toxic property of CCL4. L-Stat3−/− mice could survive (Table 1) although the hepatocyte DNA synthesis in the CCl4-treated L-Stat3−/− mice decreased as compared with their Stat3f/+ littermate controls at 40 h (Figure 5c and d) and there was no pathological abnormally in the L-Stat3−/− mice after 14 days of CCL4 injection (not shown). These data suggest that the role of STAT3 in liver mass recovery and DNA synthesis of lean mice might be ‘shrunken’ compared with those in the obese mice, which had 3- to 5-fold reduction of mitotic cells in the Alb-Cre-driven Stat3 deletion.21, 22
One possible explanation for the limited role of STAT3 in liver regeneration could be a substitution of STAT3 function by another member of the STAT family. For example, we could detect a strong activation of STAT1 in whole-liver extracts from mice lacking STAT3 in hepatocytes 3 and 8 h after PH, whereas in control mice only STAT3 was activated (Figure 6a). IL-6 is known to activate not only STAT3 but also to a lower extent STAT1. STAT1 can bind at two of the four STAT3 binding sites with a lower affinity. It is possible that in mice lacking STAT3 in hepatocytes IL-6 can activate STAT1 which substitutes for the function of STAT3 in liver regeneration. Studies using STAT1/STAT3 double-deficient mice in the liver are necessary to prove this hypothesis.
Inflammatory Infiltrates in the Regenerating Liver of L-Stat3−/− Mice
It is known that STAT3 in the immune organ is required to inhibit inflammation.36 In our study, neutrophil and monocyte infiltration was found in the liver of CCl4-treated L-Stat3−/− mice (Figure 6b). The mechanism for the increased inflammation is unclear but it seems that the inflammatory infiltrates was due to the lower level of STAT3 in the extra-hepatic organ of the L-Stat3−/− mice. These data may have clinical implication due to the fact that in a human with Stat3 gene dysfunction, which tends to be generalized and not restricted to the liver, the inflammation may even more severe because of the failure of inhibition of inflammation by STAT3 in extra-hepatic immune system.
Conclusion
Taken together, the data of the present study provide evidence that STAT3 is required for survival in the acute stage after 70% hepatectomy. After the acute stage, STAT3 plays a limited role in DNA synthesis and liver mass recovery in lean mice. In addition, exaggerated inflammatory reaction exists after hepatocyte necrosis in STAT3-deficiency mice. The recognition that STAT3 deficiency is related to the increased mortality and inflammation is expected to develop clinical management against the STAT3 inefficiency to increase survival chance in liver injury.
References
Fausto N, Campbell JS, Riehle KJ . Liver regeneration. Hepatology 2006;43 (2 Suppl 1):S45–S53.
Michalopoulos GK, DeFrances MC . Liver regeneration. Science 1997;276:60–66.
Yamada Y, Kirillova I, Peschon JJ, et al. Initiation of liver growth by tumor necrosis factor: deficient liver regeneration in mice lacking type I tumor necrosis factor receptor. Proc Natl Acad Sci USA 1997;94:1441–1446.
Cressman DE, Greenbaum LE, DeAngelis RA, et al. Liver failure and defective hepatocyte regeneration in interleukin-6- deficient mice. Science 1996;274:1379–1383.
Wuestefeld T, Klein C, Streetz KL, et al. Interleukin-6/glycoprotein 130-dependent pathways are protective during liver regeneration. J Biol Chem 2003;278:11281–11288.
Blindenbacher A, Wang X, Langer I, et al. Interleukin 6 is important for survival after partial hepatectomy in mice. Hepatology 2003;38:674–682.
Tewari M, Dobrzanski P, Mohn KL, et al. Rapid induction in regenerating liver of RL/IF-1 (an I kappa B that inhibits NF-kappa B, RelB-p50, and c-Rel-p50) and PHF, a novel kappa B site-binding complex. Mol Cell Biol 1992;12:2898–2908.
FitzGerald MJ, Webber EM, Donovan JR, et al. Rapid DNA binding by nuclear factor kappa B in hepatocytes at the start of liver regeneration. Cell Growth Differ 1995;6:417–427.
Rana B, Xie Y, Mischoulon D, et al. The DNA binding activity of C/EBP transcription factor is regulated in the G1 phase of the hepatocyte cell cycle. J Biol Chem 1995;270:18123–18132.
Greenbaum LE, Li W, Cressman DE, et al. CCAAT enhancer-binding protein beta is required for normal hepatocyte proliferation in mice after partial hepatectomy. J Clin Invest 1998;102:996–1007.
Kishimoto T . Interleukin-6: from basic science to medicine — 40 years in immunology. Annu Rev Immunol 2005;23:1–21.
Takeda K, Noguchi K, Shi W, et al. Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc Natl Acad Sci USA 1997;94:3801–3804.
Gao B . Interaction of alcohol and hepatitis viral proteins: implication in synergistic effect of alcohol drinking and viral hepatitis on liver injury. Alcohol 2002;27:69–72.
Haga S, Terui K, Zhang HQ, et al. Stat3 protects against Fas-induced liver injury by redox-dependent and -independent mechanisms. J Clin Invest 2003;112:989–998.
Alonzi T, Maritano D, Gorgoni B, et al. Essential role of STAT3 in the control of the acute-phase response as revealed by inducible gene inactivation (correction of activation) in the liver. Mol Cell Biol 2001;21:1621–1632.
Klein C, Wustefeld T, Assmus U, et al. The IL-6-gp130-STAT3 pathway in hepatocytes triggers liver protection in T cell-mediated liver injury. J Clin Invest 2005;115:860–869.
Taub R . Hepatoprotection via the IL-6/Stat3 pathway. J Clin Invest 2003;112:978–980.
Radaeva S, Sun R, Pan HN, et al. Interleukin 22 (IL-22) plays a protective role in T cell-mediated murine hepatitis: IL-22 is a survival factor for hepatocytes via STAT3 activation. Hepatology 2004;39:1332–1342.
Hong F, Jaruga B, Kim WH, et al. Opposing roles of STAT1 and STAT3 in T cell-mediated hepatitis: regulation by SOCS. J Clin Invest 2002;110:1503–1513.
Heim MH, Gamboni G, Beglinger C, et al. Specific activation of AP-1 but not Stat3 in regenerating liver in mice. Eur J Clin Invest 1997;27:948–955.
Li W, Liang X, Kellendonk C, et al. STAT3 contributes to the mitogenic response of hepatocytes during liver regeneration. J Biol Chem 2002;277:28411–28417.
Haga S, Ogawa W, Inoue H, et al. Compensatory recovery of liver mass by Akt-mediated hepatocellular hypertrophy in liver-specific STAT3-deficient mice. J Hepatol 2005;43:799–807.
Inoue H, Ogawa W, Ozaki M, et al. Role of STAT-3 in regulation of hepatic gluconeogenic genes and carbohydrate metabolism in vivo. Nat Med 2004;10:168–174.
Yang SQ, Lin HZ, Mandal AK, et al. Disrupted signaling and inhibited regeneration in obese mice with fatty livers: implications for nonalcoholic fatty liver disease pathophysiology. Hepatology 2001;34 (4 Part 1):694–706.
Selzner M, Clavien PA . Failure of regeneration of the steatotic rat liver: disruption at two different levels in the regeneration pathway. Hepatology 2000;31:35–42.
Wu H, Wade M, Krall L, et al. Targeted in vivo expression of the cyclin-dependent kinase inhibitor p21 halts hepatocyte cell-cycle progression, postnatal liver development and regeneration. Genes Dev 1996;10:245–260.
Yan C, Costa RH, Darnell Jr JE, et al. Distinct positive and negative elements control the limited hepatocyte and choroid plexus expression of transthyretin in transgenic mice. EMBO J 1990;9:869–878.
Collins FS . Positional cloning: let's not call it reverse anymore. Nat Genet 1992;1:3–6.
Radaeva S, Jaruga B, Hong F, et al. Interferon-alpha activates multiple STAT signals and down-regulates c-Met in primary human hepatocytes. Gastroenterology 2002;122:1020–1034.
Bowman T, Symonds H, Gu L, et al. Tissue-specific inactivation of p53 tumor suppression in the mouse. Genes Dev 1996;10:826–835.
Bellovino D, Morimoto T, Pisaniello A, et al. In vitro and in vivo studies on transthyretin oligomerization. Exp Cell Res 1998;243:101–112.
Soprano DR, Soprano KJ, Goodman DS . Retinol-binding protein and transthyretin mRNA levels in visceral yolk sac and liver during fetal development in the rat. Proc Natl Acad Sci USA 1986;83:7330–7334.
Makover A, Soprano DR, Wyatt ML, et al. An in situ-hybridization study of the localization of retinol-binding protein and transthyretin messenger RNAs during fetal development in the rat. Differentiation 1989;40:17–25.
Costa RH, Van Dyke TA, Yan C, et al. Similarities in transthyretin gene expression and differences in transcription factors: liver and yolk sac compared to choroid plexus. Proc Natl Acad Sci USA 1990;87:6589–6593.
Moshage H . Cytokines and the hepatic acute phase response. J Pathol 1997;181:257–266.
Akira S . Roles of STAT3 defined by tissue-specific gene targeting. Oncogene 2000;19:2607–2611.
Acknowledgements
We thank Klaus Rajewsky for providing the polβF/F mouse line, Koji Eshima for his expertise and help in the general mouse work, and Pandelakis Koni for breeding polβF/F mice. We are grateful to James Boyer for his critical evaluation of liver sections and helpful discussions. We also thank Lan Ji for technical assistance, and Thomas Welte and Nancy Bennett for critical reading of the manuscript. Y Iwamoto was supported by a fellowship of the United States Army Medical Research and Material Command (DAMD 17-97-1-7222). RA Flavell is an investigator of the Howard Hughes Medical Institute. This study was supported by a pilot project from Yale Liver Center (DK34989) and grants from the National Institutes of Health (RO1-AI34522 and RO1-AR44906) to X-Y Fu, who was also a recipient of a Career Development Award from NIH.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Moh, A., Iwamoto, Y., Chai, GX. et al. Role of STAT3 in liver regeneration: survival, DNA synthesis, inflammatory reaction and liver mass recovery. Lab Invest 87, 1018–1028 (2007). https://doi.org/10.1038/labinvest.3700630
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.3700630
Keywords
This article is cited by
-
Distinctive role of inflammation in tissue repair and regeneration
Archives of Pharmacal Research (2023)
-
gp130/STAT3 signaling is required for homeostatic proliferation and anabolism in postnatal growth plate and articular chondrocytes
Communications Biology (2022)
-
Kindlin-2 preserves integrity of the articular cartilage to protect against osteoarthritis
Nature Aging (2022)
-
Promotion of NR1I3-mediated liver growth is accompanied by STAT3 activation
Molecular Biology Reports (2022)
-
p-STAT3 is a PDC-E2 interacting partner in human cholangiocytes and hepatocytes with potential pathobiological implications
Scientific Reports (2021)
