
Abstract
Expression of Met, the Hepatocyte Growth Factor receptor, has been shown to have prognostic value in numerous types of cancer including breast, gastric, cervical and head and neck carcinomas. However, traditional analyses of expression have shown variable results and a lack of reproducibility. The AQUA® system is a method of quantitative in situ analysis of protein expression that allows the assessment of reproducibility of both antibodies and assay conditions. Here, we illustrate the necessity for antibody validation when assaying the prognostic value of a potential biomarker. Using five antibodies to the intracellular domain of the Met receptor and 10 cell line controls, we quantitatively assess reproducibility of protein expression. We show that many antibodies are not reproducible at a quantitative level from lot to lot or assay to assay, suggesting new criteria for antibody validation. We also build upon past literature addressing the prognostic value of Met in a cohort of 640 cases of invasive breast cancer on a tissue microarray. We show that high levels of expression of nuclear Met, as determined by antibodies to the intracellular domain and defined as nuclear by subcellular compartmental analysis, is associated with shorter disease-specific survival in breast cancer.
Similar content being viewed by others


Main
Met, the receptor for Hepatocyte Growth Factor (HGF), is involved in signaling pathways responsible for cell motility, migration and invasion. The receptor contains an extracellular domain that interacts with its ligand HGF, a juxtamembrane domain responsible for downregulation of Met, an intrinsic tyrosine kinase domain and a COOH-terminal docking site (reviewed by Birchmeier and Gherardi1). Upon binding to HGF, a conformational change occurs in the cytoplasmic domain allowing for ATP activation of the tyrosine kinase domain and subsequent autophosphorylation of the carboxy terminal docking site. The COOH-terminal docking site is capable of binding adaptor proteins Gab1 and Grb2, which, in turn, recruit an array of signal transducing proteins. This causes activation of a number of signaling pathways leading to cell proliferation, increased motility and branching morphogenesis, key pathways in embryonic development, epithelial to mesenchymal transitions and wound healing (reviewed by Zhang and Vande Woude2). In mammary gland development, HGF is released from the surrounding mesenchyme triggering epithelial growth and morphogenesis in epithelial cells through the Met receptor and its activation of the PI3K pathway by activating a program for ductal tree branching and inhibition of terminal differentiation.1, 3
High level expression of Met has been associated with poor prognosis in many types of cancers, including breast, gastric, cervical and head and neck carcinomas. In breast cancer, high or low expression of the Met receptor in comparison with normal epithelium has been associated with poor prognosis in lymph node negative breast carcinomas.4, 5, 6 Overexpression of Met identifies a subset of cancers independent of Her2, EGFR and other hormone receptors7 but is associated with increased Ki-67 staining.8 Such studies suggest a correlation between Met overexpression and progression of disease and metastasis. Levels of mRNA for c-Met have been positively correlated with differentiation in cancer cell lines.9 This is supported by further studies showing increasing Met expression in normal epithelium, normal epithelium within invasive carcinomas, ductal carcinoma in situ (DCIS) and highest expression in invasive carcinomas.10 These studies also point to the heterogeneity of Met found within tumors, the greatest expression being found on the ‘invasive front’ of the tumor.8, 11
Most of the studies showing a correlation between Met and disease progression have been done using the traditional immunoperoxidase/diamino-benzoate (DAB)-based staining system with antibodies to the COOH-terminus of Met. One such study found that antibodies to the intracellular domain of Met are predictive of worse outcome in lymph node negative breast carcinomas while antibodies to the extracellular domain are not predictive.5 It has been noted in some studies that immunohistochemical expression of Met is not only membranous and cytoplasmic, but also at times, strongly nuclear, particularly in the ‘invasive front’ of tumors.8 A recent study from our lab may explain this observation. We have shown that a cytoplasmic fragment of the Met receptor is generated in less differentiated cells and translocates to the nucleus; yet upon differentiation or formation of mature cell–cell contacts, these cells retain Met at the membrane.21
In this study, we assess the antibody reproducibility for many of the Met-directed antibodies used in previous studies. The highly variable results illustrate the need for antibody validation prior to protein expression analysis on valuable human tissue cohorts. Reproducibility of these COOH-terminal Met antibodies was determined using a breast carcinoma tissue microarray. Using this cohort and automated quantitative analysis (AQUA), a reproducible Met monoclonal antibody was identified and used to analyze the predictive value of Met in the nucleus. In a cohort of 640 breast carcinomas, nuclear expression of Met is predictive of worse outcome (P=0.02) in a 5-year survival analysis.
Methods
Cell Lines and Antibodies
HEK293 cells were acquired from ATCC (Manassas, VA USA) and cultured in DMEM supplemented with 10% FBS. Antibodies to the cytoplasmic domain of Met were mouse monoclonals 3D4 from Zymed Laboratories (San Francisco, CA, USA) and MAB3729 from Chemicon (Temecula, CA, USA). Polyclonal antibodies were C12 and C28 from Santa Cruz Biotechnologies (Santa Cruz, CA, USA) and CVD13 from Zymed. DO24 is a mouse monoclonal antibody from Upstate (Lake Placid, NY, USA) directed to the extracellular domain of Met.
Western Blot Analysis
HEK293 cells were harvested in RIPA buffer. Lysate concentrations were measured using BioRad reagent and 40 μg of total lysate was loaded onto 10% SDS-polyacrylamide gels and transferred to Hybond ECL nitrocellulose membrane (Amersham Biosciences, Piscataway, NJ, USA). Membranes were blocked in 1% bovine serum albumin, Tris-buffered saline and 1% Tween (BSA/TBS/Tween) for 1 h. Primary antibodies at a 1:500 dilution were added to BSA/TBS/Tween and incubated for 1 h at room temperature. Membranes were washed with TBS/Tween and then incubated with horseradish peroxidase-conjugated goat anti-mouse or goat anti-rabbit secondary antibody (Jackson Labs, Bar Harbor, ME, USA) at a dilution of 1:20 000 for 1 h at room temperature. Signals were detected using ECL reagent (Amersham Biosciences, Piscataway, NJ, USA) and exposed to the film.
Tissue Microarray Construction
Tissue microarrays were constructed from formalin-fixed, paraffin-embedded breast cancer specimens as described previously.12 Tumor regions were selected by pathologists for coring. The tissue microarray was constructed with 0.6-mm-diameter cores spaced 0.8 mm apart using a Tissue Microarrayer (Beecher Instruments, Sun Prairie, WI, USA). The tissue microarray block was cut into 5 μm sections, adhered to the slide by an adhesive tape-transfer method (Instrumedics Inc., Hackensack, NJ, USA) and UV crosslinked.
Patient Cohort
The breast cohort consists of 688 samples of invasive ductal carcinoma serially collected from the Yale University Department of Pathology from 1961 to 1983. The mean follow-up time is 12.8 years with a mean age of diagnosis of 58.1 years. This cohort contains approximately half node-positive and half node-negative specimens. Detailed treatment information was not available for this cohort.
Immunohistochemistry
Tissue microarrays were deparaffinized by two xylene rinses followed by 100% and 70% ethanol washes. Antigen retrieval was performed by pressure cooking slides in sodium citrate (pH 6.0). Slides were incubated for 30 min in 2.5% hydrogen peroxide in methanol to block endogenous peroxidase activity followed by 0.3% bovine serum albumin in 1 × TBS for 1 h to reduce non-specific background staining. Slides were incubated first with an anti-cytokeratin monoclonal or polyclonal antibody (DAKO, Carpinteria, CA, USA) and a Met antibody overnight at 4°C. Slides were washed three times in TBS and incubated with secondary antibodies Alexa 488 (Molecular Probes, Eugene, OR, USA) and mouse or rabbit Envision (DAKO, Carpinteria, CA, USA). The slides were washed three times in TBS described above and incubated for 10 min with Cy-5 tyramide (NEN Life Science Products, Boston, MA, USA). The slides were then mounted in Prolong Gold with DAPI (Molecular Probes, Eugene, OR, USA) and covered with a cover slip.
Automated Quantitative Analysis of Protein Expression
Tissue microarray images were taken as part of an automated image acquisition and analysis as described previously.13 Briefly, images of tissue and cell line cores were captured with an Olympus BX51 microscope and analyzed by Automated Quantitative Analysis (AQUA™). For each core, areas of tumor are distinguished from stroma by creating an epithelial tumor mask created from the cytokeratin signal. The tumor mask was determined by binary gating of the cytokeratin pixels. The DAPI image, which was used to identify the nuclear compartment, was subjected to a rapid exponential subtraction algorithm improving signal-to-noise ratio by subtracting the out-of-focus image from the in-focus image. After application of the rapid exponential subtraction algorithm, the signal intensity of the target protein (eg Met) was scored on a scale of 0–255. The AQUA score within subcellular compartments is calculated by dividing the signal intensity by the area of the specified compartment or the entire tumor mask. The AQUA technique has been extensively described in previous works12, 13 and is now documented in over 40 peer-reviewed publications.
Statistical Analysis
Statistical analysis of regressions and Cox proportional hazards was done using JMP 5.0.1 (SAS Insititute, Cary, NC, USA). The cohort was split into randomized training and validation sets using X-tile14 as a method for selecting optimal cut-points.
Results
Validation of Met Antibodies by Western Blot
As an initial analysis of each antibody, Western blot analysis of HEK293 lysates was performed. For this study, we assessed the monoclonal antibodies 3D4 and MAB3729, with epitopes directed at the tyrosine kinase domain of Met, and polyclonal antibodies C12, C28 and CVD13, directed at the COOH-terminal docking site of Met, and DO24, a monoclonal antibody to the extracellular domain. HEK293 cells were selected for analysis since they express the 60 kDa nuclear fragment of Met at levels that allow for detection by Western blotting in the absence of cell treatment (as described previously21), even though they express very low levels of the full length Met receptor. Figure 1 shows all intracellular domain antibodies detecting the 145 kDa Met receptor as well as the 60 kDa nuclear fragment of Met with the exception of MAB3729, which did not detect full-length Met under these conditions and DO24, which has been previously shown to be unable to detect full-length Met on a Western blot.

Met antibody validation on Western blot. (a) HEK293 cells express low levels of the Met receptor and its 60 kDa fragment. Antibodies 3D4, C12, CVD13 and C28 detect the Met receptor (145 kDa) and the 60 kDa fragment of Met in HEK293 cell lysate. MAB3729 detects the 60 kDa fragment but not the full length receptor under these conditions. 3D4 and C28 detect other bands that are known truncation forms of the Met receptor. The antibody DO24, directed at the extracellular domain of Met, does not detect Met on Western blots and is not shown. (b) Antibody epitopes to the Met receptor are depicted on the schematic representing the Met receptor.
Lot to Lot Variability of Antibodies
To assess antibody reproducibility, tissue arrays containing 640 breast cancers were stained with each antibody using different TMA masterblocks from the same cohort or different sections from the same TMA masterblock. As seen in Figure 2, a high degree of variability was observed on slides stained with different lots of the same antibody. In Figure 2a, 3D4, a mouse monoclonal antibody shows clear nuclear expression of Met in a histospot representative of the typical expression pattern for the majority of the cohort. A second lot of the antibody on a histospot from the same tumor from the same patient shows a very different staining pattern in Figure 2b. This staining appears more membranous and non-specific and is again representative of the majority of the histospots. The variability between tumor spots from the same tumor from a single patient (redundant histospots) on the two arrays is shown in Figure 2c as a scatterplot with an R-value of 0.194. However, both lots of the 3D4 antibody showed similar profiles on Western blots of HEK293 cell lysate. In comparison, another monoclonal antibody (MAB3729) showed a correlation of R=0.57 between redundant histospots (Figure 2d) and a correlation of R=0.80 in deeper sections of the same histospot (Figure 2e). These values are more typical of our experience using the AQUA system in our lab on either redundant core or deeper section tissues from the same patient. The degree of variability seen in 3D4 led us to examine a range of commercially available Met antibodies on multiple tissue array slides from the same patient cohort to attempt to define a standard for validation.

Variability between lots of antibodies. Monoclonal antibody 3D4 shows variation in staining patterns from lot to lot. Although both samples of antibody detected full-length Met on a Western blot consistently (data not shown), Lot 1 clearly shows a nuclear staining pattern (a), whereas Lot 2 shows a membranous or cytoplasmic staining pattern (b) on breast tissue samples. Staining was carried out on two slides from the same array, and although these are not the same tumor spot, the staining pattern pictured here is representative of the expression patterns seen on the entire slide. This is an example of an obvious case of irreproducibility; other lots of antibodies were not nearly as different to the eye but only detectable by quantitative analysis by a computer. Variability as detected on scatterplots of redundant histospots on two different arrays using different lots of 3D4 (c) or MAB3729 (d). Scatterplot e shows variability between deeper cuts from the same array, using different lots of antibody MAB3729.
One of Five Met Antibodies is Reproducible by Quantitative analysis
Although tissue arrays have been extensively validated as a method to assess protein expression in large cohorts15, 16 there is inherent variability in protein expression from array to array of the same cohort due to biological tissue/tumor heterogeneity. Thus, it cannot be assumed that qualitative analysis of protein expression between TMA would correlate perfectly. We assess correlation between two slides from the same cohort by linear regression analysis. An example of a linear regression analysis is shown in Figure 2c–e. Studies from our lab (unpublished data) have previously shown regression values of 0.9 or better between slides cut from the same masterblock (deeper sections) and values of 0.70 or better between arrays of the same cohort for traditional well-established biomarkers such as PR, ER and Her2. However, in our study of these five Met antibodies, regression analyses between arrays stained with the same Met antibody have values less than 0.4 as shown in Table 1a. This suggests that these antibodies are not providing a reproducible assessment of Met protein expression when analyzed quantitatively. Furthermore, the regression profiles between the antibodies against different regions of the Met protein show poor correlation, even assessed on deeper sections in the same arrays (Table 1b). Two antibodies that show a good correlation between arrays are MAB3729, a mouse monoclonal antibody from Chemicon directed at the tyrosine kinase domain and C28, a polyclonal antibody from Santa Cruz directed at the last 28 amino acids in the COOH-terminal tail. MAB3729 gives an R-value of 0.6 between arrays (Figure 2d) and 0.8 (Figure 2e) on slides of the same array. These R-values suggest somewhat more variability than we have seen for our traditional biomarkers, which may be explained by greater tumor heterogeneity of the Met protein. To confirm this, we looked at cell line controls on each array. The R-value between the cell lines on masterblocks 49_4 and 49_5 for MAB3729 was 0.94, well within our acceptable range. This confirms that the antibody is indeed reproducible between arrays of the same cohort and is consistent with the smaller R-values illustrating tumor heterogeneity. In addition, results from MAB3729 and C28 antibodies on the same array correlated well with an R-value of 0.71. Neither of these antibodies correlated with the results from antibodies C12, CVD13 or 3D4. However, a second lot of C28 was not reproducible. The antibody required a different dilution for optimal staining and resulted in an R-value of 0.29 between 49_4 and 49_5. Furthermore, the example in Figure 2 would suggest that the difference in antibody lots is detectable by eye. In this study, all other antibodies tested showed similar patterns of membranous, cytoplasmic and nuclear expression of Met, with membranous expression being observed the least.
Met is Predictive of Worse Outcome in Breast Carcinoma
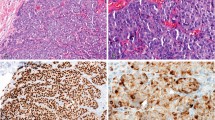
Once we identified an antibody that is reproducible for quantitative analysis (MAB3729), we were able to analyze the data collected. Owing to the greater amount of heterogeneity seen by Met in tumors, two slides from the same array (12 cuts or about 60 μm apart) and one slide from a second array were used. AQUA was performed on images from the breast cohort as illustrated in Figure 3. The resulting AQUA scores were divided by the exposure time to obtain a score relative to 1 ms of exposure time in order to normalize for subtle changes in exposure time due to the age of the light source and variables related to fluorophore yield. These normalized scores were averaged and used for statistical analysis. X-Tile14 is a statistical method to determine the optimal cut-point for a continuous population. It was used to find the best cut-point for a training set derived from a randomized selection of half of the population. As AQUA scores provide continuous data, rather than the ordinal data from traditional pathologist-read IHC, we are unable to use cut-points defined in previous publications. Figure 4a shows an X-tile plot. It depicts the possible cut-points for the training set on a triangular grid, as described in the study of Camp et al.14 The intensity and color of each cut-point on the X-tile plot represents the direction and significance of that cut-point in relation to patient outcome. The red color on the X-tile plot indicates that the low population of nuclear Met expressing patients has better outcome at many different cut-points. The optimal cut-point for the training set was 0.4 AQUA units per millisecond. This cut-point was then applied to the second half of the randomized population, the validation set. Using this cut-point, Kaplan–Meier analysis was carried out on both the training and validation sets (Figure 4b and d) resulting in a validation set log rank P-value of 0.02 indicating that patients with high nuclear Met expression have a significantly worse outcome at 5 years. Specifically, Figure 4 shows high nuclear Met has a 65% 5-year survival compared with a 75% 5-year survival for the low expression group.

Automated Quantitative Analysis (AQUA) of Met antibody MAB3729. (a) A representative tissue spot stained with cytokeratin is used to identify areas of tumor to create a tumor mask. (b) The tumor mask is compartmentalized into nuclear (blue) and non-nuclear (green) regions based on the location of DAPI staining within the tumor mask (c) Met expression (d) is placed within a compartment and the intensity of expression is given an AQUA score on a continuous scale of 0–255.

X-tile plot of Met nuclear expression, optimal cut-point selection by training and validation set. X-tile plot representing all possible cut-points for the training set (a) and validation set (c) of nuclear Met data. Each colored pixel is a single cut-point where patient number increases down the plot for high expressers or to the right for low expressers. The intensity and color of the pixel represents the direction of the relationship with patient survival. (Red represents better outcome for the lower population.) Histograms depict the population distribution in the training set (b) and validation set (d) with their Kaplan–Meier plots as determined using the X-tile optimal cut-point of 0.4. Red represents low expressers and green represents high expressers.
Univariate analysis was done using Cox proportional hazards model. Univariate analysis shows a significant relationship between patient survival and nuclear expression of Met with a hazard ratio of 1.66 and a P-value of 0.0289, as shown in Table 2. Other variables showing a significant relationship with patient survival are nodal status (P<0.0001), nuclear grade (P=0.0017), tumor size (P<0.0001), high ER (P<0.0001), high PR (P<0.001) and high Her2 (P=0.0083). Age and histologic grade showed no relationship with patient survival.
Multivariate analysis on the validation set of the cohort was completed with a Cox proportional hazards model using the markers from Table 2 that show significance with outcome. Table 3a shows that nodal status, tumor size, ER and Her2 status are independently predictive of outcome in this model. Multivariate analysis on a model including ER, PR, Her2 and nuclear Met indicate that nuclear expression of Met is a predictor of worse outcome (P=0.0206) identifying a novel subset of cancers independent of traditional biomarkers ER, PR and Her2. Addition of Tumor Size to this model as shown in Table 3d, indicates that nuclear Met remains an independent marker. Addition of Nodal Status to the model shown in Table 3b results in P=0.0512 (Table 3c), indicating that nuclear Met as a marker has a slight relationship with nodal status. Overall, these results indicate that nuclear expression of Met can identify a subset of cancers independent of ER, PR, Her2 and tumor size, but may have a relationship with nodal status in this patient cohort. This is consistent with previous studies where subcellular localization was not addressed.6, 17, 18, 19
Discussion
The exact required methods for antibody validation are not well established. However, the validation step is critically important to insure accuracy of immunohistochemical analysis.20 Here, we have shown that four of five commercially available antibodies to the Met COOH-terminal domain are not reproducible at a quantitative level even though most of them accurately identify the target protein on a Western blot. Although we identified MAB3729 as a reproducible antibody, which shows results highly similar to some lots of C28 antibody, it is clear that there are many sources for non-reproducibility. During this study, lots of MAB3729 were seen to lose their specificity after 4–5 months of proper storage. The best polyclonal antibody, C28, showed that a new lot required a new antibody titer and although it appeared qualitatively very similar, it was not rigorously reproducible.
Another variable to take into consideration when validating an antibody for quantitative analysis is the degree of heterogeneity. It has been reported that there is a higher degree of heterogeneity of Met expression in tumors than has been seen in proteins such as ER and PR. Through regression analysis of cell lines, we have confirmed this observation as the cell line controls on different arrays correlated well with R-values of 0.9 or better when using the antibody MAB3729. However, the tumor spots on these same arrays had much lower R-values, between 0.5 and 0.6, pointing to greater heterogeneity in Met expression within a tumor or possibly other artifacts present in tissue that are not present in cell lines.
All attempts to assess protein expression in situ are fraught with many uncontrollable variables. TMAs represent a valuable method to minimize some of those variables. However, antibody selection remains as a critical variable. This study was able to identify one out of five antibodies to the same protein that gives a reproducible result at a quantitative level. The antibody most commonly used in the literature (C28) showed promise and looked qualitatively similar. But quantitative analysis of each lot showed relatively poor reproducibility (one lot was reproducible, one was not). This illustrates the importance of quantitative validation of protein expression, even on a lot to lot basis. If these antibodies were to be used as a companion diagnostics, it would be critical to include reproducibility standards. The findings that many of these antibodies are not reproducible does not necessarily suggest that past immunohistochemical studies on Met are all inaccurate or incorrect. Most of the literature on Met as a prognostic marker has been done using traditional IHC methods (ie brown staining) with scoring by a pathologist. Frequently those studies were not specific with respect to the subcellular localization of the expression pattern. With the exception of 3D4, we were only able to detect irreproducibility between antibodies when the intensity of the staining pattern was determined through a highly sensitive automated system for quantitative analysis. AQUA enhances our ability to detect differences in protein expression by scoring the staining intensity on a continuous scale of 0–255, increasing the sensitivity of the assay in comparison with traditional scoring systems of 0–3. However, in light of these findings, antibodies should be carefully validated for reproducibility even if they are not being used with an automated system as there can be visible lot to lot differences as was seen with the antibody 3D4.
Our previous studies have shown that cytoplasmic domain antibodies to Met are predictive for survival whereas extracellular domain antibodies are not predictive.19 That study used traditional IHC-based analysis on a cohort that is a subset of the cases used for this current study. Thus, above AQUA-based results are confirmatory and consistent with results obtained by traditional methods.19 However, in that study, no attempt was made to assess subcellular localization. A number of years after completion of that study, we have shown that a fragment of Met containing the cytoplasmic domain can be found in the nucleus of less differentiated cells, whereas mature or differentiated cells retain Met at the plasma membrane.21 Here, the finding that nuclear expression of Met, as detected by antibodies to the cytoplasmic domain, is predictive of worse outcome in a 5-year survival analysis and is consistent with the biological finding that nuclear expression is seen in low-density cultures and at the edge of wounded epithelial sheets in wound healing type assays (Pozner-Moulis and Rimm, unpublished observations). This raises the possibility that nuclear Met identifies a subpopulation of aggressive breast carcinomas independent of the traditional biomarkers ER, PR and Her2, which may be similar to the wound healing signature class identified by molecular profiling by the Stanford group.22
References
Birchmeier C, Gherardi E . Developmental roles of HGF/SF and its receptor, the c-Met tyrosine kinase. Trends Cell Biol 1998;8:404–410.
Zhang YW, Vande Woude GF . HGF/SF-met signaling in the control of branching morphogenesis and invasion. J Cell Biochem 2003;88:408–417.
Yang Y, Spitzer E, Meyer D, et al. Sequential requirement of hepatocyte growth factor and neuregulin in the morphogenesis and differentiation of the mammary gland. J Cell Biol 1995;131:215–226.
Camp RL, Rimm EB, Rimm DL . Met expression is associated with poor outcome in patients with axillary lymph node negative breast carcinoma. Cancer 1999;86:2259–2265.
Kang JY, Dolled-Filhart M, Ocal IT, et al. Tissue microarray analysis of hepatocyte growth factor/Met pathway components reveals a role for Met, matriptase, and hepatocyte growth factor activator inhibitor 1 in the progression of node-negative breast cancer. Cancer Res 2003;63:1101–1105.
Tsarfaty I, Alvord WG, Resau JH, et al. Alteration of Met protooncogene product expression and prognosis in breast carcinomas. Anal Quant Cytol Histol 1999;21:397–408.
Tolgay Ocal I, Dolled-Filhart M, D'Aquila TG, et al. Tissue microarray-based studies of patients with lymph node negative breast carcinoma show that met expression is associated with worse outcome but is not correlated with epidermal growth factor family receptors. Cancer 2003;97:1841–1848.
Edakuni G, Sasatomi E, Satoh T, et al. Expression of the hepatocyte growth factor/c-Met pathway is increased at the cancer front in breast carcinoma. Pathol Int 2001;51:172–178.
Cheng HL, Trink B, Tzai TS, et al. Overexpression of c-met as a prognostic indicator for transitional cell carcinoma of the urinary bladder: a comparison with p53 nuclear accumulation. J Clin Oncol 2002;20:1544–1550.
Jin L, Fuchs A, Schnitt SJ, et al. Expression of scatter factor and c-met receptor in benign and malignant breast tissue. Cancer 1997;79:749–760.
Beviglia L, Matsumoto K, Lin CS, et al. Expression of the c-Met/HGF receptor in human breast carcinoma: correlation with tumor progression. Int J Cancer 1997;74:301–309.
Dolled-Filhart M, McCabe A, Giltnane J, et al. Quantitative in situ analysis of {beta}-catenin expression in breast cancer shows decreased expression is associated with poor outcome. Cancer Res 2006;66:5487–5494.
Camp RL, Chung GG, Rimm DL . Automated subcellular localization and quantification of protein expression in tissue microarrays. Nat Med 2002;8:1323–1327.
Camp RL, Dolled-Filhart M, Rimm DL . X-tile: a new bio-informatics tool for biomarker assessment and outcome-based cut-point optimization. Clin Cancer Res 2004;10:7252–7259.
Camp RL, Charette LA, Rimm DL . Validation of tissue microarray technology in breast carcinoma. Lab Invest 2000;80:1943–1949.
Torhorst J, Bucher C, Kononen J, et al. Tissue microarrays for rapid linking of molecular changes to clinical endpoints. Am J Pathol 2001;159:2249–2256.
Ghoussoub RAD, Dillon DA, Daquila T, et al. Expression of C-Met is a strong independent prognostic factor in breast carcinoma. Cancer 1998;82:1513–1520.
Edakuni G, Sasatomi E, Satoh T, et al. Expression of the hepatocyte growth factor/c-Met pathway is increased at the cancer front in breast carcinoma. Pathol Int 2001;51:172–178.
Kang JY, Dolled-Filhart M, Ocal IT, et al. Tissue microarray analysis of hepatocyte growth factor/met pathway components reveals a role for met, matriptase, and hepatocyte growth factor activator inhibitor 1 in the progression of node-negative breast cancer. Cancer Res 2003;63:1101–1105.
McShane LM, Altman DG, Sauerbrei W, et al. Reporting recommendations for tumor marker prognostic studies (REMARK). J Natl Cancer Inst 2005;97:1180–1184.
Pozner-Moulis S, Pappas DJ, Rimm DL . Met, the hepatocyte growth factor receptor, localizes to the nucleus in cells at low density. Cancer Res 2006;66:7976–7982.
Chang HY, Nuyten DS, Sneddon JB, et al. Robustness, scalability, and integration of a wound-response gene expression signature in predicting breast cancer survival. Proc Natl Acad Sci USA 2005;102:3738–3743.
Acknowledgements
We thank Jennifer Giltnane and Mark Gustavson for their indispensable knowledge and help. This work was supported in part by the Department of Defense Breast Cancer Research Program Grant No. W81XWH-04-1-0438 (SPM) and by NIH R33 CA 110511 (DLR).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Pozner-Moulis, S., Cregger, M., Camp, R. et al. Antibody validation by quantitative analysis of protein expression using expression of Met in breast cancer as a model. Lab Invest 87, 251–260 (2007). https://doi.org/10.1038/labinvest.3700515
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.3700515
Keywords
This article is cited by
-
Development of [89Zr]ZrDFO-amivantamab bispecific to EGFR and c-MET for PET imaging of triple-negative breast cancer
European Journal of Nuclear Medicine and Molecular Imaging (2021)
-
MET canonical transcript expression is a predictive biomarker for chemo-sensitivity to MET-inhibitors in hepatocellular carcinoma cell lines
Journal of Cancer Research and Clinical Oncology (2021)
-
Data integration from pathology slides for quantitative imaging of multiple cell types within the tumor immune cell infiltrate
Diagnostic Pathology (2017)
-
Pathological examination of breast cancer biomarkers: current status in Japan
Breast Cancer (2016)
-
The clinical and functional significance of c-Met in breast cancer: a review
Breast Cancer Research (2015)
