
Abstract
Quantitative reverse transcriptase real-time PCR (QRT-PCR) is a robust method to quantitate RNA abundance. The procedure is highly sensitive and reproducible as long as the initial RNA is intact. However, breaks in the RNA due to chemical or enzymatic cleavage may reduce the number of RNA molecules that contain intact amplicons. As a consequence, the number of molecules available for amplification decreases. We determined the relation between RNA fragmentation and threshold values (Ct values) in subsequent QRT-PCR for four genes in an experimental model of intact and partially hydrolyzed RNA derived from a cell line and we describe the relation between RNA integrity, amplicon size and Ct values in this biologically homogenous system. We demonstrate that degradation-related shifts of Ct values can be compensated by calculating delta Ct values between test genes and the mean values of several control genes. These delta Ct values are less sensitive to fragmentation of the RNA and are unaffected by varying amounts of input RNA. The feasibility of the procedure was demonstrated by comparing Ct values from a larger panel of genes in intact and in partially degraded RNA. We compared Ct values from intact RNA derived from well-preserved tumor material and from fragmented RNA derived from formalin-fixed, paraffin-embedded (FFPE) samples of the same tumors. We demonstrate that the relative abundance of gene expression can be based on FFPE material even when the amount of RNA in the sample and the extent of fragmentation are not known.
Similar content being viewed by others


Main
Prognostic factors in cancer are mainly clinical and histopathological parameters and molecular analyses based on measurements of expression levels are not yet widely used. However, this situation may change dramatically when the power of expression level measurements becomes more generally accepted and when these techniques can be applied routinely. The existence of characteristic gene expression profiles has recently been documented with high-density DNA chips for several tumor subtypes in breast and other types of cancer.1, 2, 3, 4, 5, 6, 7, 8 For example, primary breast cancer in 295 patients was tested with high-density cDNA chips representing 25 000 genes. A set of 70 genes was characterized, which was sufficient to discriminate between primary breast cancer, which has a good prognosis, and cancer, which tends to have bad prognosis.4, 6 This classification system was shown to outperform all clinical variables in predicting the likelihood of distant metastases to appear within 5 years. Discrimination on the basis of gene expression profiles can therefore be a strong and independent factor for predicting the outcome of breast cancer. While it was very important to analyze the expression of thousands of genes to identify those genes that contribute to a classification of tumors into distinct subgroups, the measurement of the relatively few prognostically relevant genes would be sufficient to establish a prognostic tool.
Archival material is stored in all pathological institutes for many years, but unfortunately as formalin-fixed, paraffin-embedded (FFPE) tissue blocks and not as frozen samples. While numerous reports documented the feasibility of reverse transcription followed by quantitative PCR of RNA derived from frozen sections, analyses based on FFPE material are much more demanding.9, 10, 11, 12, 13, 14, 15, 16, 17 A major challenge relates to the sometimes extensive degradation of RNA due to the fixation procedure (paraformaldehyde),18 which may even become worse when sections are incubated in aqueous solution; for example, during additional immunohistochemical stainings.19 Large batteries of archival samples are stored as FFPE material in many pathological institutes; therefore, a methodology based on such samples would be of great value and might facilitate the discovery and validation of genes whose expression correlates with clinically relevant subcategories of tumors.
DNA chip-based studies are highly sensitive to RNA degradation. In contrast, quantitative reverse transcriptase real-time PCR (QRT-PCR) is more robust and tolerates a partial degradation of RNA. However, extensive RNA degradation leads to a loss of amplifiable templates in the RNA population, which is paralleled by a loss of amplicons and an increase in threshold values (Ct values) in subsequent QRT-PCR. This is especially critical for RNA derived from FFPE tissue, where fragments are in the range of less than 100 to about 250 nucleotides.
We developed a procedure that allows to compensate for the effect of RNA degradation on subsequent expression measurements by reverse transcription of RNA followed by quantitative real-time PCR, and we show that reliable results can be obtained with RNA derived from FFPE samples.
Materials and methods
Cell Cultures and Tissue Specimens
ZR-75-1 (ATCC number CRL-1500) is a breast cancer cell line originally isolated from a metastatic site of a 63-year-old woman. Cells were grown in RPMI containing 10% fetal calf serum (Sigma) at 37°C in 5% CO2.
Tumor samples were provided from the Tumorbank Bern (Switzerland) following the guidelines of the Bernese Ethics Committee. Fresh samples were immersed in RNAlater (Qiagen) or processed for histological evaluation by standard FFPE. Fixation was done in 4% paraformaldehyde in PBS. Buffered formalin was used to prevent acidification of the tissue during fixation.
RNA Extraction and Chemical Degradation
Cultured cells were harvested in PBS, washed and resuspended in RNA extraction buffer (RLT) and homogenized with a polytrone. RNA was extracted by selective binding to a silica gel-based membrane (RNeasy, Qiagen). Four 25 μm thick sections were prepared from RNAlater-preserved tumor specimen at −30°C, homogenized in RLT with a polytrone and RNA was extracted according to the RNeasy protocol (Qiagen). A different protocol was used to prepare RNA from FFPE samples: ten 20 μm thick sections were deparaffinized by extracting twice in xylene for 20 min at 37°C and dehydrated in 99% RNase free ethanol and air dried. Sections were lysed in 650 μl chaotropic 2 × Nc RNA Lysis Solution, (Applied Biosystems, Foster City, CA, USA) and digested for 12–16 h at 55°C in the presence of 500 μg proteinase K.12, 13, 20 RNA was purified on an ABI PRISM™ 6100 Nucleic Acid PrepStation (Applied Biosystems) and collected in 100 μl water or elution solution. RNA was quantitated spectroscopically (NanoDrop, Witec, Switzerland) and quality was assessed by capillary electrophoresis (Agilent 2100 Bioanalyzer, Agilent Technologies, Palo Alto, CA, USA).
The ZR-75-1 RNA was partially hydrolyzed by incubation in a 0.5 M NaHCO3/Na2CO3 buffer (pH=10) for 1.5–120 min at 60°C, as indicated in the figures. Hydrolysis of RNA was stopped by the addition of 3 M NaAc pH 5.2 and precipitation with ethanol (100%). RNA degradation was assessed by capillary electrophoresis using a Bioanalyzer and RNA 6000 ladder as reference RNA (Ambion, Nr. 7152).
Primer/Probe Design for TaqMan Assay PCR
The nucleotide sequences and exon–intron boundaries of GAPDH (hCG2005673), IGFBP5 (hCG16384), ERBB2 mRNA (hCG28177) and 18S ribosomal RNA (X03205) were derived from the Celera Discovery Systems™ online platform (https://myscience.appliedbiosystems.com) or NCBI (http://www3.ncbi.nlm.nih.gov/). Primer and probes were designed with Primer Express® Software v. 2.0 (Applied Biosystems, Foster City, CA, USA). Probes were labeled with 6-FAM™ at the 5′ end and with a nonfluorescent MGB quencher at the 3′ end. The forward primer and probe for each gene was combined with two or three different reverse primers resulting in short amplicons (54–65 bp), medium-size amplicons (83–111 bp) and long amplicons (103–147 bp) (Table 1). Primers and probes were from Applied Biosystems (Warrington, UK).
Reverse Transcription and Expression Analysis
Reverse transcription of RNA derived from FFPE and RNAlater material was performed from 250 ng of total RNA and 125 U Multiscribe™ reverse transcriptase (High-Capacity cDNA Archive Kit, Applied Biosystems, Foster City, CA, USA). Random prime cDNA was synthesized from intact and hydrolyzed ZR-75-1 RNA with AMV reverse transcriptase, following the protocol of the manufacturer (Roche diagnostics). Expression was measured from 5 ng of cDNA per reaction with primer probe sets representing short, medium-size and long amplicons (Table 1). Real-time PCR was also carried out from RNA which was not reverse transcribed (no RT control). Each cDNA was then analyzed on TaqMan® Low Density Arrays (Applied Biosystems). These arrays are prefabricated 384-well cards, where each well is spotted with gene-specific primers and probes during manufacturing. Arrays with four sets of 96 genes were used. Each reaction is carried out with 0.5–1 ng of mRNA/cDNA per well in a final volume of 1 μl. Primer/probe sets were chosen from the large collection of TaqMan® Gene Expression Assays, with optimized primer and probe sets (Applied Biosystems, Foster City, CA, USA). Where possible, TaqMan gene expression assays with probes spanning exon–exon boundaries were chosen in order to minimize signals from contaminating chromosomal DNA. The list of Assays is given in Table 2. cDNAs were mixed with 2 × TaqMan® Universal Mastermix (Applied Biosystems) and 100 μl were loaded into each fill port on the Low Density Arrays. In all, 48 wells on the array are connected to a fill port through microchannels. cDNAs are distributed into the wells by a short centrifugation. Channels were sealed as recommended by the manufacturer and QRT-PCR was performed on an Applied Biosystems 7900HT instrument.
Normalization of Data
Absolute threshold cycle values (Ct values) were determined with the SDS software v. 2.1 (Applied Biosystems). Expression data of 15 putative control genes were included on each set of 96 genes. Those control genes that had the lowest variation between the 15 tumors were identified with GeNorm, a software which was developed to determine the optimal control genes from a larger list of putative control genes, which showed the lowest variation among tumors in all the tumors.21
Immunohistochemistry
Immunohistochemistry was carried out according to standard protocols and essentially as described previously.22
Results
QRT-PCR in Intact and Partially Hydrolyzed RNA from ZR-75-1 Cells
We used a well-defined test system to establish the correlation between RNA fragmentation and QRT-PCR. The system is based on gene-specific primers and 6-FAM™ labeled TaqMan® probes specific for four genes and RNA derived from ZR-75-1 breast cancer cells. The RNA was subjected to a controlled chemical hydrolysis, resulting in a highly reproducible size distribution of RNA depending on the time of hydrolysis. We then determined Ct values by QRT-PCR reactions using oligonucleotides coding for short, medium-size and long amplicons.
QRT-PCR is dependent on two primers flanking the amplicon and an internal, fluorescently labeled probe. Degradation of RNA results in a loss of intact amplicons in the RNA and consequently in cDNA, thereby leading to an increase in Ct values. We first demonstrated that short amplicons are more robust against RNA fragmentation than longer amplicons. Intact RNA was partially hydrolyzed by incubating for increasing time at elevated pH and temperature. By using the same batch of RNA, any biological variation could be eliminated. Aliquots of fragmented RNA were subjected to electrophoretic separation on a Bioanalyzer to define the extent of hydrolysis and size distribution of fragments. Substantial fragmentation and disappearance of 28S ribosomal RNA was observed within 7.5 min of hydrolysis. At about 30 min, RNA was degraded to fragments of about 200 nucleotides and hydrolysis went on to fragments of 75–85 nucleotides after 120 min (Figure 1a). Also shown is a representative RNA sample derived from a FFPE sample of breast cancer. Degradation of RNA was also measured in cryo-sections that were fixed with formalin and treated for 0.25–16 h with paraffin at 62°C (Figure 1b). Figure 1c shows electropherograms of RNA samples from Figure 1a. Figure 1d summarizes graphically the correlation between time of hydrolysis and relative size of fragmented RNA for three independent analyses. Each RNA sample was subjected to QRT-PCR using forward primers and probes specific for 18S ribosomal RNA, GAPDH, Insulin-like growth factor binding protein (IGFBP5) and ERBB2, which were combined with various reverse primers resulting in short, medium-size and long amplicons ranging from 54 to 147 bp (Table 1). Figure 1e summarizes the result of amplification of chemically hydrolyzed RNA with three amplicons of different sizes for GAPDH and Figure 1f shows Ct values of short, medium-size and long amplicons for 18S RNA with paraffin-treated samples. QRT-PCR with short and medium-size amplicons of GAPDH coding for 54 and 83 bp long amplicons were similarly efficient, while a more pronounced increase of Ct values was observed with primers coding for the long amplicon (103 bp). Expression values for all four genes were determined at each time point of chemical fragmentation and a strong negative dependence between RNA fragmentation and raw expression values was observed for all four genes tested. This experiment was performed with three separate batches of RNA and three independent series of hydrolysis. Each batch was analyzed separately with all the primer/probe sets for the four genes. Figure 1e shows mean expression values for GAPDH. In this experimental system, the increase of Ct values reflects the loss of amplicons due to the fragmentation of RNA.

Threshold cycles (Ct) values in real-time PCR correlate with RNA integrity. (a) RNA was isolated from ZR-75-1 human breast cancer cells and subjected to partial hydrolysis by incubation for up to 120 min at pH 10 and 60°C as indicated. RNA fragmentation was determined by capillary electrophoresis (Agilent Bioanalyzer) using an RNA ladder as size marker (size of fragments: 0.2, 0.5, 1.0, 2.0, 4.0 and 6.0 kb). (b) Four 20 μm thick sections were fixed with formalin and treated with paraffin for 0.25, 2, 6 and 16 h before isolation of total RNA. Electropherograms corresponding to intact RNA and RNA hydrolyzed for 7.5, 30, 60 and 120 min, and RNA derived from a FFPE sample are shown in panel (c). The relation between the time of hydrolysis and the size of the resulting RNA fragments was determined for three independent batches of RNA and mean values and s.d. are shown (bold line) (d). Relative expression values were determined with primers encoding short (open circles), medium-size (open squares) and long amplicons (open triangles) of GAPDH. Shown are mean Ct values of GAPDH for all three series of ZR-75-1 RNA along with s.d. (e). Panel (f) shows Ct values of 18S RNA prepared from fixed material after treatment with paraffin for 0 to 16 h.
This system of chemically hydrolyzed RNA derived of ZR-75-1 RNA was used to demonstrate the power of normalization. Expression levels of three test genes were determined with probe sets detecting amplicons of various size. Each expression value was normalized against the mean Ct value of the three amplicons specific for 18S RNA. As there is no biological variation between different time points of hydrolysis in this test system, the selection of the control gene was not critical in this experiment. We measured Ct values of three mRNAs (GAPDH, IGFBP5 and ERBB2) with short, medium-size and long amplicons and determined relative Ct values to 18S ribosomal RNA (delta Ct=Cttest gene− mean Ct18S). Expression values were determined in intact RNA and in RNA after 1.5, 3, 7.5, 15, 30, 60, 80, 100 and 120 min of hydrolysis (Figure 2a–c). As the absolute Ct values increased similarly for both the test and the control gene, fairly constant delta Ct values were observed for all three test genes in this system even in heavily degraded RNA (Figure 2a–c). It is also apparent that delta Ct values were more constant with short amplicons (triangles) than with medium-size (circles) or long amplicons (squares). The experiment was carried out with three independent RNA preparations.

Relative gene expression levels of three test genes (IGFBP5, GAPDH, ERBB2) were determined from intact and partially hydrolyzed ZR-75-1 RNA. RNA was hydrolyzed by incubation at elevated pH for 1.5–120 min (see Materials and methods). Shown are delta Ct values (between each test gene and 18S ribosomal RNA). All determinations were performed from three independently prepared and hydrolyzed RNA isolations. The cDNAs derived from 18S RNA were diluted 1:1000-fold before QRT-PCR.
It was of interest to investigate whether a similar system of normalization could be applied to a larger set of genes and commercially available primer/probe assays. The RNA expression levels were measured on TaqMan® Low Density Arrays. We used Low Density Arrays that contained primer/probe-combinations for 96 genes. Selection criteria were based on DNA chip studies on breast cancer and on data based on immunohistochemical studies. In all, 67 genes were expressed at a measurable level in ZR-75-1 cells. Figure 3a shows that Ct values again increased with fragmentation of the RNA. This increase was very similar for the great majority of genes including 18S RNA (lowest Ct values, ≈13 at time 0 and ≈17 at time 120 min). The resulting correlation coefficients between Ct values and time (from 0 to 80 min) were greater than 0.80 (P<0.024) for all but three genes and greater than 0.95 for 54 genes (P<0.003) (Figure 3b). This result suggests that many genes are similarly degraded in this system. Included on the Low Density Arrays were 15 putative control genes (Table 2) and four of them were selected as control genes by applying the procedure of Vandesompele et al.21 They were used to compensate for the changes in expression levels resulting from increasing fragmentation of RNA by hydrolysis. Delta Ct values (delta Ct=Cttest gene− Ctmean of four control genes) were determined for each gene on the Low Density Arrays and for each time point of hydrolysis. Figure 3c shows that delta Ct values remained rather constant even at advanced fragmentation of the RNA. This suggests that this normalization procedure is suitable to efficiently normalize expression levels for weakly and extensively fragmented RNA.

Ct values correlate with the extent of RNA fragmentation. Ct values were determined by QRT-PCR on TaqMan® Low Density Arrays for a panel of 67 genes including control genes (a). Pearson's correlations between Ct values and time of hydrolysis (0 and 80 min) are shown for all the 67 genes (b). Delta Ct values were determined at each time point of hydrolysis as the difference between the Ct value for each gene and the mean Ct of four control genes (c).
Expression Measurements in Human Breast Cancer Samples
We investigated whether this procedure of compensation could be applied to RNA that was degraded during fixation in formalin and embedding in paraffin. Fresh frozen tumor material of 15 human breast cancers was analyzed in parallel to FFPE material of the same tumors. Total RNA was isolated from four 25 μm thick RNAlater-preserved cryo-sections and from ten 20 μm thick paraffin sections of each tumor. RNA isolated from RNAlater-preserved samples was of high quality (28S/18S ratio >1.5), RNA isolated from FFPE material was of poor quality and the distribution of fragments was similar to ZR-75-1 RNA hydrolyzed in vitro for 30 to 60 min (see lane ‘FFPE’, Figure 1a).
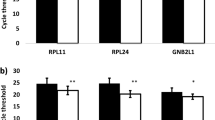
The Ct values were measured for all 96 genes on the Low Density Arrays in all 15 RNAlater-preserved and matched FFPE samples and the results were visualized separately for each tumor as boxplots (Figure 4). The median Ct values were on average 6 units higher in RNA derived from FFPE material than in RNA derived from fresh frozen samples. In contrast to the test system, not only experimental variations but also biological variations between independent samples must be leveled out with these samples before the results can be compared. Therefore, 15 putative control genes were again included on the Arrays, from which we expected rather constant expression levels. The most stably expressed control genes were selected according to the procedure previously described by Vandesompele et al.21 Briefly, two or more endogenous control genes should be very similarly expressed in all the samples, while the absolute level of expression of a control gene is not critical. Proper control genes must not necessarily parallel absolute amounts of RNA as this value is predominantly dictated by ribosomal RNA. Vandesompele proposed an s.d. of log-transformed control gene ratios as a robust measure for the variation between two control genes because this measure is independent of the absolute abundance of any control gene. Using this procedure, we determined four appropriate control genes based on RNA derived from the 15 RNAlater-preserved samples (SDHA, 18S, RPLP0, HPRT1). We assessed whether FFPE material would nevertheless allow relative measurements of expression levels. Delta Ct values were compared for 60 genes between RNAlater- and FFPE-derived samples. A mean correlation coefficient of 0.79±0.046 (s.d.; minimum 0.69, maximum 0.84) was found between matched Ct values in the 15 tumors. Figure 5a shows the result for one representative tumor (C1). Similarly, correlation coefficients were determined separately for each gene on the card and the result for GAPDH is shown (Figure 5b). Correlation coefficients for this comparison were generally lower. This may be due to the fact that only 15 pairs of RNAlater-preserved and FFPE-treated samples were available for this comparison.

Comparison of QRT-PCR results derived from high- and poor-quality RNA. Total RNA was isolated from RNAlater-preserved and from FFPE sections and processed for QRT-PCR with Low Density Arrays. Ct values were determined for each gene in each of the 15 tumors and values were normalized based on four control genes (SDHA, 18S rRNA, RPLP0, HPRT1) as described in the text. Shown are boxplots of normalized Ct values of RNAs isolated from RNAlater-preserved material and RNA derived from FFPE material. Ct values greater than 40 were excluded from the evaluation.

Correlation between raw Ct values derived from RNAlater-preserved and FFPE-derived material. Shown is the result of tumor C1. Pearson's correlation between matched Ct values of the two samples was 0.77 (a). Panel (b) shows the correlation between raw Ct values derived from RNAlater-preserved and FFPE-derived material for GAPDH (Pearson's correlation 0.6).
The Ct values determined from FFPE and RNAlater-preserved material were further tested for the estrogen receptor (ER), a clinically important parameter. which is routinely determined by immunohistochemistry with an ER-specific antibody. An ER 1-specific probe was included on the Low Density Arrays and the ER expression was measured in high-quality RNA and in RNA derived from paraffin sections and compared with the immunohistochemical data of the same tumors (Figure 6). Tumors were arranged along the x-axis according to increasing ER levels determined semiquantitatively by immunohistochemistry. A good coincidence was observed between protein and normalized RNA data in all the 15 samples tested. In two tumors that expressed very low (C9) or no ER on the basis of immunohistochemical staining (C15), no signals were detected in RNA derived from FFPE samples.

Measurement of ER levels by semiquantitative immunohistochemistry and QRT-PCR. Tumors C1–C15 were sorted along the x-axis according to increasing ER levels as determined semiquantitatively by immunohistochemistry. Average ER levels are plotted as a solid line (0–100%, right-hand scale). ER levels were determined by QRT-PCR on Low Density Arrays from RNAlater-preserved tumor material (hatched boxes) and from FFPE material (gray boxes) and expression levels are plotted in logarithmic scale for each tumor. FFPE material from C15 and C9 revealed nonmeasurable QRT-PCR values.
Discussion
An increasing number of reports mainly based on DNA chip studies demonstrate that expression profiles or gene signatures may be very powerful to separate tumors into clinically relevant subtypes that cannot be discriminated so far by classical methods. For example, it is well known that some patients with node-negative primary breast cancer can be cured by surgery alone, while tumors reappear in another group of patients. There is currently no classical way to discriminate between the two groups. As a consequence, many patients undergo adjuvant therapy although only some actually profit from it. Similarly, the outcome cannot be predicted reliably in most cases of primary breast cancer. Both parameters depend on long follow-up periods and therefore, assays based on archival material, ideally derived of clinically controlled studies, would be optimal for the search and validation of critical genes or gene expression profiles. For most studies, such samples are only available as FFPE samples, and therefore, a method based on such material might be of great impact. We and others demonstrate that RNA isolated from FFPE material can be used for quantitative gene expression measurements.10, 11, 12, 15, 16, 17, 23, 24, 25 The fact that this method is still not widely used may reflect that several parameters affect the performance of critical steps of the procedure. This includes the efficiency of RNA recovery from archival samples, fragmentation of the RNA during fixation, quality of PCR primers and size of the amplified amplicon. Some of these steps are quite difficult to control. While many problems related to the isolation of RNA from FFPE tissue could be solved with high concentration of proteinase K and long incubation times at elevated temperature, the fragmentation of the RNA seems to remain the most critical factor limiting the procedure. We analyzed in more detail the consequences of RNA fragmentation and amplicon size on RT-PCR and Ct values. Already, from a theoretical point of view, it can be postulated that QRT-PCR is affected by RNA fragmentation, as every break in the RNA that occurs between two primers inevitably separates the two ends of the amplicon into two different cDNA molecules and is therefore lost as template for subsequent PCR. Therefore, short amplicons should be used for quantitation (Figure 2).14, 16, 26 We used a complex RNA derived from a human cell line to measure the relation between RNA fragmentation and threshold cycle values (Ct values) in QRT-PCR reactions and we demonstrate that even small differences in amplicon size are critical, especially for extensively degraded RNA. By using homogeneous pools of intact starting RNA, the complexity of fragmented RNA was identical to the complexity of starting RNA, resulting in a system that was free of biological variation. Using this system we demonstrate that Ct values strongly depend on the RNA integrity and Ct values increase when RNA is degraded (Figure 1d). This is true not only for massively fragmented RNA (hydrolyzed for more than 60 min) but also for RNA that is only moderately degraded (Figure 1d, 15 min of hydrolysis or less). Fragmentation of RNA during formalin-fixation leads to a partial hydrolysis of RNA during the fixation and embedding process. Once the tissue is fixed, dehydrated and embedded in paraffin, further degradation of RNA is very limited or stops.
Degradation after partial hydrolysis at elevated pH leads to a broad peak of RNA fragments where the median size of fragmented RNA depends on the time of hydrolysis: at 15 min of hydrolysis the peak is around 500 nucleotides, after 30 min it is at 200 and after 60 min it is at around 100 nucleotides. Interestingly, similar distributions of RNA fragments were observed after chemical hydrolysis or after fixation and embedding in paraffin (Figure 1a and b) and also after limited cleavage with RNase A in vitro (data not shown).27 RNA derived from FFPE is heavily degraded to fragments in the range of less than 100 nucleotides to about 200 nucleotides. Expression measurements from such a fragmented RNA are paralleled by a similar increase of Ct values in subsequent RT-PCR analyses (Figure 1e and f).
As there was no biological variation in RNA from ZR-75-1 cells, the selection of control genes was not critical. Calculation of relative expression levels (difference between Ct of gene of interest minus Ct of ribosomal RNA, delta Ct values) almost completely levels off the fragmentation-mediated increase of Ct values, leading to rather stable delta Ct values irrespective of the extent of fragmentation. It is also apparent that small amplicons result in more stable delta Ct values than relatively large amplicons (Figure 2). The same phenomenon was previously shown for large amplicons which failed to generate measurable expression values.14, 16, 26
We also tested fragmented RNA from ZR-75-1 cells with a panel of genes arranged on Low Density Arrays. Increasingly hydrolyzed RNA resulted in very similar increases of Ct values of most genes on the card (Figure 3a and b). Rather stable delta Ct values were determined as the difference between the Ct value of each test gene minus the mean of four control genes for each point of fragmentation (Figure 3c). The results strongly suggest that fragmentation-induced changes in Ct values can be compensated.
This procedure was then applied to RNAs derived from 15 human breast cancer samples. RNA was prepared from well-preserved tissue and from FFPE samples. As expected the absolute Ct values determined from RNA derived of FFPE material were higher than those of RNA derived of fresh tissue (Figure 4). Expression values were normalized based on four control genes that were determined from a panel of 15 potential control genes according to the procedure described by Vandesompele et al.21 We compared delta Ct values from RNA of fresh material and RNA of FFPE material from each tumor and a mean correlation coefficient of 0.77±0.04 (s.d.; minimum 0.69, maximum 0.87) was found between Ct values determined from RNA of fresh frozen and of FFPE samples (Figure 5a). In addition, we compared expression levels of each gene between high- and poor quality RNA in all 15 tumors (Figure 5b). This comparison allows one to examine each primer/probe set individually and the resulting correlation coefficient is a good measure of the quality of each primer/probe set.
It is known that progression of many breast cancers which overexpress the ER can be inhibited by antiestrogens or drugs, that inhibit the production of endogenous estrogens. We compared the expression of the ER at the RNA level with the ER protein as measured by immunohistochemistry (Figure 6) and found a good accordance between the two measurements. Tumors which had a low level of ER protein also expressed low ER mRNA levels. Similar results were obtained with RNA data derived of good-quality (from RNAlater) and poor-quality RNA (from FFPE samples).
Our results demonstrate that archival breast cancer samples may be used for expression profiling. RNA derived from FFPE archival samples will always be substantially degraded and this inevitably affects accuracy and sensitivity of expression measurements based on real-time PCR. Nevertheless, reliable measurements can still be made for many genes.
References
Davis RE, Staudt LM . Molecular diagnosis of lymphoid malignancies by gene expression profiling. Curr Opin Hematol 2002;9:333–338.
Sotiriou C, Neo SY, McShane LM, et al. Breast cancer classification and prognosis based on gene expression profiles from a population-based study. Proc Natl Acad Sci USA 2003;100:10393–10398.
Sorlie T, Perou CM, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA 2001;98:10869–10874.
van de Vijver MJ, He YD, van’t Veer LJ, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med 2002;347:1999–2009.
Ramaswamy S, Tamayo P, Rifkin R, et al. Multiclass cancer diagnosis using tumor gene expression signatures. Proc Natl Acad Sci USA 2001;98:15149–15154.
van’t Veer LJ, Dai H, van de Vijver MJ, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002;415:530–536.
Huang E, Cheng SH, Dressman H, et al. Gene expression predictors of breast cancer outcomes. Lancet 2003;361:1590–1596.
Chang JC, Wooten EC, Tsimelzon A, et al. Gene expression profiling for the prediction of therapeutic response to docetaxel in patients with breast cancer. Lancet 2003;362:362–369.
Rupp GM, Locker J . Purification and analysis of RNA from paraffin-embedded tissues. Biotechniques 1988;6:56–60.
Reichmuth C, Markus MA, Hillemanns M, et al. The diagnostic potential of the chromosome translocation t(2;13) in rhabdomyosarcoma: a Pcr study of fresh-frozen and paraffin-embedded tumour samples. J Pathol 1996;180:50–57.
Sheils OM, Sweeney EC . TSH receptor status of thyroid neoplasms—TaqMan RT-PCR analysis of archival material. J Pathol 1999;188:87–92.
Abrahamsen HN, Steiniche T, Nexo E, et al. Towards quantitative mRNA analysis in paraffin-embedded tissues using real-time reverse transcriptase-polymerase chain reaction: a methodological study on lymph nodes from melanoma patients. J Mol Diagn 2003;5:34–41.
Cronin M, Pho M, Dutta D, et al. Measurement of gene expression in archival paraffin-embedded tissues: development and performance of a 92-Gene Reverse Transcriptase-Polymerase Chain Reaction Assay. Am J Pathol 2004;164:35–42.
Specht K, Richter T, Muller U, et al. Quantitative gene expression analysis in microdissected archival formalin-fixed and paraffin-embedded tumor tissue. Am J Pathol 2001;158:419–429.
Stanta G, Bonin S . RNA quantitative analysis from fixed and paraffin-embedded tissues: membrane hybridization and capillary electrophoresis. Biotechniques 1998;24:271–276.
Godfrey TE, Kim SH, Chavira M, et al. Quantitative mRNA expression analysis from formalin-fixed, paraffin-embedded tissues using 5′ nuclease quantitative reverse transcription-polymerase chain reaction. J Mol Diagn 2000;2:84–91.
Finke J, Fritzen R, Ternes P, et al. An improved strategy and a useful housekeeping gene for RNA analysis from formalin-fixed, paraffin-embedded tissues by PCR. Biotechniques 1993;14:448–453.
Mizuno T, Nagamura H, Iwamoto KS, et al. RNA from decades-old archival tissue blocks for retrospective studies. Diagn Mol Pathol 1998;7:202–208.
Fend F, Emmert-Buck MR, Chuaqui R, et al. Immuno-LCM: laser capture microdissection of immunostained frozen sections for mRNA analysis. Am J Pathol 1999;154:61–66.
Masuda N, Ohnishi T, Kawamoto S, et al. Analysis of chemical modification of RNA from formalin-fixed samples and optimization of molecular biology applications for such samples. Nucleic Acids Res 1999;27:4436–4443.
Vandesompele J, De Preter K, Pattyn F, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 2002;3:1–12 (RESEARCH0034).
Baltzer A, Svanborg C, Jaggi R . Apoptotic cell death in the lactating mammary gland is enhanced by a folding variant of alpha-lactalbumin. Cell Mol Life Sci 2004;61:1221–1228.
Lewis F, Maughan NJ, Smith V, et al. Unlocking the archive—gene expression in paraffin-embedded tissue. J Pathol 2001;195:66–71.
D’Orazio D, Stumm M, Sieber C . Accurate gene expression measurement in formalin-fixed and paraffin-embedded tumor tissue. Am J Pathol 2002;160:383–384.
Specht K, Richter T, Muller U, et al. Quantitative gene expression analysis in microdissected archival tissue by real-time RT-PCR. J Mol Med 2000;78:B27.
Lehmann U, Kreipe H . Real-time PCR analysis of DNA and RNA extracted from formalin-fixed and paraffin-embedded biopsies. Methods 2001;25:409–418.
Kuschel M, Ausserer W . Characterization lf RNA quality using the Agilent 2100 Bioanalyzer. Agilent Technologies Application Notes 2000.
Acknowledgements
This work was supported by the Swiss National Science Foundation (Grant 31-61929 to RJ, and NCCR ‘Plant Survival’ to DRG), the Bernese Cancer League and by Applied Biosystems, Rotkreuz, Switzerland. Human material was provided by the Tumorbank Bern. The Tumorbank Bern is sponsored by the Department of Clinical Research, the Institute of Pathology of the University of Bern and the Bernese Cancer League.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Antonov, J., Goldstein, D., Oberli, A. et al. Reliable gene expression measurements from degraded RNA by quantitative real-time PCR depend on short amplicons and a proper normalization. Lab Invest 85, 1040–1050 (2005). https://doi.org/10.1038/labinvest.3700303
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.3700303
Keywords
This article is cited by
-
Recommendations for mRNA analysis of micro-dissected glomerular tufts from paraffin-embedded human kidney biopsy samples
BMC Molecular Biology (2018)
-
Systematic evaluation of RNA quality, microarray data reliability and pathway analysis in fresh, fresh frozen and formalin-fixed paraffin-embedded tissue samples
Scientific Reports (2018)
-
Straightforward and sensitive RT-qPCR based gene expression analysis of FFPE samples
Scientific Reports (2016)
-
Validation of adequate endogenous reference genes for reverse transcription-qPCR studies in human post-mortem brain tissue of SIDS cases
Forensic Science, Medicine, and Pathology (2015)
-
Quantitative assessment of short amplicons in FFPE-derived long-chain RNA
Scientific Reports (2014)
