
Abstract
HIV viral protein R (Vpr) affects the immunocyte cell cycle and circulates as free polypeptide in plasma of AIDS patients. Effects of Vpr on cardiomyocytes were explored using transgenic mice (TG) with Vpr targeted to cardiomyocytes by the α-myosin heavy-chain promoter. TG and WT littermate hearts were evaluated histopathologically, ultrastructurally, molecularly via RNA microarray analysis and quantitative RT-PCR, and functionally by cardiac magnetic resonance imaging (MRI) and electrocardiograms (ECG). Six hemizygous lines were created (Vpra,b,c,d,e,h). Vpr RNA was expressed exclusively in myocardium and Vpr mRNA expression correlated with phenotypic changes. Vprb exhibited the highest expression and mortality. TGs developed congestive heart failure (≈8 weeks), abnormal cardiomyocyte nuclei and mitoses (≈12 weeks), and became moribund (≈20 weeks) with atrial mesenchymal tumors. MRI revealed four-chamber dilation, defective contraction, and atrial masses. Pathologically, cardiomegaly and atrial mesenchymal tumors occurred (≈16–20 weeks). ECGs showed prolonged R–R, Q–T, and P–R intervals (≈12 weeks). RNA encoding collagen and bone morphogenic protein 4, 6, and 7 were increased. Vpr targeted to cardiomyocytes caused defective contractility and atrial tumors. Since some Vpr cardiomyocytic effects resemble those found in terminally differentiated immunocytes, some pathogenetic mechanisms may be shared at the subcellular level.
Similar content being viewed by others

Main
Viral protein R (Vpr) is an HIV accessory gene that encodes a unique 96-residue polypeptide required in macrophage infection.1 Early in the epidemic, extracellular Vpr was found at high levels in serum of AIDS patients,2 and reflected disease. Since Vpr is cell permeable, it is reasonable to suggest that Vpr could have functional implications in diverse cell types,3 including terminally differentiated cardiomyocytes.4 In mononuclear cells, Vpr causes G2/M cell cycle arrest and apoptosis.5 Conversely, mutant Vpr (R77Q) is associated with nonprogressive HIV infection and impaired apoptosis.6 Vpr was implicated in the development of congestive heart failure (CHF) and dysrhythmias in AIDS,7 but a mechanism to explain the clinical observation was lacking.
A cardiac-targeted Vpr transgene (TG) (driven by the α-MyHC promoter) offered an approach to dissect the effects of this important HIV gene product on the structure and function of the heart without the systemic effects of AIDS. Six TG lines were created. All developed atrial cardiomyocyte proliferation with aberrant mitoses and defects in cardiomyocyte nuclear structure (Vprb developed them at ≈8–12 weeks), atrial mesenchymal tumors, cardiac dysrhythmias, and CHF. The phenotype worsened temporally and with gene dosage. The distinctive pathological and physiological phenotype and altered gene expression in vivo recapitulated important Vpr effects on terminally differentiated immunocytes.8, 9 Findings here may have implications in the pathogenesis of AIDS cardiomyopathy (CM).
Materials and methods
Generation of α-MyHC/Vpr TG Mice
Established methods were employed as described previously. The α-MyHC promoter is an extensively used,10 robust and cardiac-specific promoter that exhibits excellent tissue specificity and minimal positional effects of insertion.11 A 291-bp fragment containing the Vpr gene was amplified from HIV-1 pNL43 using the primers VprXhoI and VprH3 and cloned into a pBluescript 5K vector. The α-MyHC clone 26 (compliments of Jeff Robbins, Children's Hospital Research Foundation, Cincinnati, OH, USA12) was digested with SalI and HindIII (Roche Applied Science, Indianapolis, IN, USA) to facilitate construction of the final vector. Restriction analysis and DNA sequencing verified the final construct, denoted as α-MyHC/Vpr. To generate TGs, a 6.4-kb BamHI–BamHI fragment containing the full transcriptional unit was purified and microinjected into FVB one-cell embryos (Charles River, Wilmington, MA, USA). Embryos were implanted into pseudo-pregnant CD-1 (Taconic, Germantown, NY, USA) females. The resulting offspring were screened for incorporation of the transgene.
As controls for the specificity of Vpr effects on cardiomyocytes and the heart, cardiac targeting of HIV Vpu and Nef was accomplished using α-MyHC/Nef and /Vpu constructs, respectively, in different TG experiments. The resulting Nef and Vpu TG lines were screened for incorporation of the targeted HIV transgene construct (Nef or Vpu) as described below. In all experiments, mice were housed according to NIH guidelines and treated according to IACUC-approved protocols. Mice were watered and fed ad libitum.
Genotyping
For the TG line α-MyHC/Vpr, the TG was detected in the founders and their offspring using Southern blotting and PCR essentially as described previously for the Tat TG.13 For Southern blotting, 10 μg of mouse genomic tail DNA was digested overnight at 37°C. The digested DNA was subjected to electrophoresis in a 0.7% agarose gel and transferred to a positively charged nylon membrane (Schleicher & Schuell, Keene, NH, USA) overnight. Hybridization was performed in a solution containing 6 × SSC (1 × SSC:150 mmol/l NaCl, 15 mmol/l Na citrate), 1% SDS, 10% dextran sulfate, 100 μg/ml salmon sperm DNA, and 32P-labeled Vpr cDNA at 68°C overnight. The membranes were then washed once in 2 × SSC-1% SDS at room temperature for 15 min and twice in 0.1 × SSC–0.1% SDS at 68°C for 30 min and exposed to a storage phosphor screen (Packard Instrument, Meriden, CT, USA). Detection was performed on a Cyclone storage phosphor system (Packard).
Genotyping of the progeny from the founders was accomplished by using PCR. In all, 50 ng of mouse genomic tail DNA was used along with specific primers that were designed to detect the presence of each targeted TG. The primers used were VPR1 and VPR2, which produced a product of 291 bp. PCR was performed in a 20-μl volume using Taq DNA Polymerase (Invitrogen, Carlsbad, CA, USA) in a PTC100 (MJ Research, Watertown, MA, USA) thermal cycler under the following conditions: 5 min at 95°C followed by 30 cycles of 30 s at 95°C, 30 s at 55°C, 1 min at 72°C, ending with a 3 min extension at 72°C and then 4°C.
For the control TG lines (α-MyHC/Nef, α-MyHC/Vpu), the TG was detected in the founders and their offspring using Southern blot and PCR essentially as described by us in the recent past. Genotyping of these progeny from the founders also was accomplished by using PCR. In all, 50 ng of mouse genomic tail DNA was used along with specific primers that were designed to detect the presence of each targeted TG. The primers used were VPU1 (5′-ACGCGTCGACAATGCAACCTATAATAG-3′) and VPU2 (5′-CCCAAGCTTACTACAGATCATCAATATC C-3′) for Vpu TGs, which produced a product of 250 bp. NS1 (5′-ACGCGTCGACGATGGGTGGCAAG TG-3′) and NH3 (5′-CCCAAGCTTGTCAGCAGTTCC TGAAG-3′) for Nef, which produced a 620 bp product. PCR was performed as described above.
RNA Extraction and Northern Analysis of Vpr in the Myocardium
Methods resembled those used by us previously.13 Total RNA was extracted from mouse tissues with TRI reagent (Molecular Research, Cincinnati, OH, USA). RNA (10 μg) was subjected to electrophoresis with the NorthernMax kit (Ambion, Austin, TX, USA) in a 1% agarose gel and transferred to a positively charged nylon membrane (Roche Applied Science) overnight. Hybridization was performed in ULTRAhyb (Ambion) containing either 32P-labeled Vpr cDNA or 32P-labeled glyceraldehyde-3-phosphate dehydrogenase (GAPDH) cDNA transcripts at 42°C overnight. The membranes were then washed twice in 2 × SSC–0.1% SDS at 42°C for 5 min and twice in 0.1 × SSC–0.1% SDS at 42°C for 15 min and exposed to a storage phosphor screen (Packard). Detection was performed as above on a storage phosphor system (Packard).
For determinations of steady-state abundance of Vpr message in heart tissue, mRNA was isolated from total cardiac RNA samples with the Poly(A) Pure kit (Ambion). A measure of 2 μg of mRNA was subjected to electrophoresis as above and hybridized with probes for Vpr and GAPDH.
DNA Microarray Analysis
DNA microarray analyses (n=6 each Vprb and WT) were performed with Amersham Codelink murine bioarrays containing 10 000 genes, according to the protocols provided by the manufacturer (Amersham Biosciences, Piscataway, NJ, USA). Briefly, total RNA was extracted using RNeasy Fibrous Tissue Mini columns (QIAGEN, Valencia, CA, USA). First-strand cDNA was prepared, labeled, and processed as described in the CodeLink system protocols. Processed arrays were scanned using a GenePix Scanner (Axon Instruments, Foster City, CA, USA), and array images were acquired and analyzed using the CodeLink™ Expression 2 Software (Amersham).
Quantitative Real-Time RT-PCR of Vpr in Myocardium from Different TG Lines and of Other Target Genes from Microarray
For relative abundance of Vpr mRNA in heart tissue, mRNA was isolated from total cardiac RNA samples with the Micro Fast Track Kit (Invitrogen) and subjected to RT-PCR as described below and compared to the signal of the housekeeper GAPDH.
For confirmation of microarray data, real-time RT-PCR was performed on selected gene products to confirm the results. Total RNA (4 μg) was reverse-transcribed by using random primers and a Superscript-II kit (Invitrogen) to synthesize first-strand cDNA. The cDNA was purified and stored at −20°C until used. The cDNA was amplified using a LightCycler (Roche Applied Science) RT-PCR machine, using the FastStart DNA Master SYBR Green kit (Roche Applied Science). Relative quantitation of mRNA levels was determined using LightCycler analysis software (Roche Applied Science), which compared the amount of expression of a gene of interest to GAPDH among different samples, allowing for relative quantification of gene expression levels.
Magnetic Resonance Image of the Heart
WT (n=2) and Vprb TGs (n=3) were evaluated via magnetic resonance image (MRI) at 16–20 weeks. MRI was accomplished on a 4.7T Varian/INOVA (200/33) Spectroscopy and Imaging System (Varian, Palo Alto, CA, USA). A gradient insert with an inner diameter of 11.7 cm was used. This gradient set has a maximum gradient strength of (25 G/cm) with a rise time of 238 ms. Animal temperature and heart rate (HR) were stabilized and scout images were obtained to localize the heart. One scout image was planned with the slice plane along the long axis of the heart. From this image, a short axis R wave-triggered CINE image with 10 frames per TR was obtained with a slice position just below the heart valve of the LV. The four-chamber slice was planned from this short axis slice. Both four chamber and short axis views for functional analysis were performed. CINE images at 10 frames per TR with TR set slightly less than the period of the HR were then obtained. The field of view was 8 cm × 4 cm with 256 × 128 pixels. The data were zero filled to 512 × 256 and processed to give a final in-plane resolution of 156 × 156 mm. Two averages were obtained with a slice thickness of 1.0 mm. The pulse sequence used was a cardiac gated 2D-gradient-echo sequence with a TE of 3.2 ms. TR was set slightly less than the period of the heartbeat. TR, therefore, varied between 0.113 and 0.100 s. The rf power of the excitation pulse was optimized to give good contrast between chamber blood and myocardium. No flow compensation was used in the initial images obtained.
Real-Time Electrocardiogram in TGs
Methods employed resemble those described by Tom Hampton and co-workers,14, 15 and those we adapted and employed in the recent past.16 Briefly, mice were gently removed from their cages and positioned on the platform for electrocardiogram (ECG) recording (AnonyMOUSE™, Mouse Specifics, Boston, MA, USA). An array of gel-coated ECG electrodes are embedded in the floor of the platform and spaced to provide contact between electrodes and animals' paws. Electrodes were connected to an amplifier by shielded conductive leads. Each mouse was permitted to acclimate for 10 min before collection of baseline ECG. Anesthesia was not used. Signals were digitized (16-bit precision, 2000 samples/s). Data from continuous recordings of 15–25 ECG signals were used for analyses and interpretation.
ECG Analyses
Vpra,b,h (n=4, 16, 4, respectively; each line evaluated at 12 weeks old) were evaluated using real-time ECGs (without confounding effects of anesthesia). Each signal was analyzed using e-MOUSE, Internet-based physiologic waveform analyses software as described previously.14, 15 The program employed uses Fourier analyses and linear time-invariant digital filtering of frequencies below 2 Hz and above 100 Hz to minimize noise. Software uses a peak detection algorithm to find the peak of the R waves and to calculate HR. The inverted and/or biphasic portions of the T wave were used to calculate the Q–T interval,17 which was rate corrected (Q-Tc) by an algorithm developed for mice. Spurious data resulting from unfiltered noise or motion artifacts were rejected. The mean of the ECG time intervals for each set of waveforms was calculated. Heart rate variability (HRV) was calculated as the standard deviation of all R–R intervals for each set of ECG signals, and the coefficient of variation (CV, %) was calculated as the ratio of mean HRV to mean HR.18
Histological Sectioning and Pathological Evaluation of Hearts in WT and Vpr TG
Vpra,b,h TG hearts were evaluated histopathologically (n=4, 8, 4, respectively; 12–16 weeks old). Histological studies were accomplished in ways that resemble those used by us in the past.19 At the selected termination points, hearts were rapidly removed and fixed by immersion into neutral-buffered formalin (10%, Fisher Scientific, Pittsburgh, PA, USA). For histopathological evaluation, each heart was divided evenly with a single edge razor blade to reveal both ventricular and both atrial cavities in a single section. Both bivalved sections were processed routinely, dehydrated through a graded series of alcohols, and paraffin embedded as performed previously.19 Serial sections from the paraffin-embedded tissue slice (6 μm) were stained with hematoxylin and eosin (for cellular detail) and with Masson's trichrome and elastica von Giessen (to highlight collagen and elastic fibers, respectively). Two observers examined each slide. Photomicrographs were taken using a Nikon E800 microscope, × 0.5 lens (Nikon, Garden City, NY, USA), and with higher power apo-planachromat lenses to highlight cytologic details. For evaluation of Nef and Vpu TG hearts, methods employed were essentially identical to those described above.
Transmission Electron Microscopy
Fine structure of tumor cells Vprb, (n=3; 16–20 weeks old) were evaluated using electron microscopy (EM). For ultrastructure, sections of atrium with gross evidence of tumor were rapidly fixed in diluted Karnovsky's fixative as we have peformed in the past and processed for EM.19 Transmission EM sections were viewed on a JEOL 100CX microscope (JEOLCO, Tokyo) and evaluated by blinded observers and electronphotomicrographs taken of representative areas of the mass. For evaluation of Nef and Vpu TG cardiac ultrastructure, methods employed were essentially identical.
Statistical Analysis
For physiological, pathological, and molecular studies, groups were compared by ANOVA, essentially as we have performed in the past.20 Significance was established with P<0.05.
Results
General
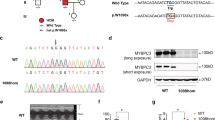
Six hemizygous TG lines were created. Vpr RNA was present exclusively in myocardium of TGs and highly expressed in Vprb. Other lines (Vpra,c,d,e,h) exhibited lower Vpr expression. Northern signal for Vpr was absent in other TG tissues including quadriceps femoris, brain, lung, liver, kidney, and spleen (Figure 1a). Using polA+ separation methods, Vpr messenger RNA was identified in heart samples from the TG (Figure 1b).

(a) Northern blot probed with Vpr cDNA in tissue samples from Vprb TG: Vpr RNA signal was present in the myocardium of TGs. Northern signal for Vpr was absent in other TG tissues including skeletal muscle (Q. femoris), brain, lung, liver, kidney, spleen. (b) PolyA+ RNA blot from Vpr TG and WT heart demonstrates abundant Vpr mRNA in the TG heart. (c) Quantitative RT-PCR analysis of Vpr expression in heart extracts from Vpra,b,h lines. Vpr expression was normalized to GAPDH. Highest Vpr expression was found with Vprb.
Quantitative RT-PCR revealed that the abundance of the mRNA was greatest in hearts from Vprb (≈20 copies) followed by Vprh (≈10 copies) and Vpra (≈4 copies; Figure 1c) indicating a gene:dose effect of the Vpr construct on cardiac structure and function.
Phenotypic changes in Vpr TGs followed the pattern of mRNA expression (above). The pathological phenotype in Vprb was robust (Figure 2a; compare TG to WT). CHF, edema, and increased body weight were evident at ≈8–12 weeks. This progressed rapidly over ≈8 weeks, and was terminal at ≈16–24 weeks. CHF was less prominent in lower expressor TGs (Vpra,c,d,e,h) where edema, anasarca, and labored respiration did not present until ≈24 weeks of age.

(a) Gross photographs of Vprb (TG) and WT littermate: Vprb exhibits profound CHF compared to WT littermate: Vprb (TG) mouse with massive effusions, ascites and edema from CHF. (b) Autopsy reveals cardiomegaly (circle) with CHF (fluid) draining from abdominal and chest cavities. (c) Kaplan–Meier survival data from the cohort of Vprb TGs and WT littermates showing premature mortality (mean survival=194d) in TG compared to WT.
Overall, the consistent pathophysiological phenotype of Vpr targeted to myocardium included CHF, atrial enlargement (unilateral or bilateral), biventricular dilation, and unilateral or bilateral atrial masses that were histopathologically defined as mesenchymal tumors. Associated with CHF in Vprb TGs, clinical and pathological features included enlarged cardiac silhouette on roentgenogram (not shown), ascites, pleural effusions, and anasarca (documented postmortem; Figure 2b).
TG lines Vpra survived longer as shown in the Kaplan–Meier survival curves (Figure 2c). Survival data for Vprb TGs (n=36) and WT littermates (n=43) were obtained and plotted (Figure 2c) with mean Vprb TG survival of 205d.
Pathological Features from Vpr TG Autopsies
Histopathological evidence of CHF included pulmonary edema and hepatic congestion (not shown). Histopathological examination of the bivalved heart in Vprb TGs (≈16–20 weeks) revealed relatively preserved ventricular structure, ventricular dilation and myocyte hypertrophy, bilateral atrial wall thinning, and easily discerned, acellular, atrial intracavitary tumors (Figure 3a). This acellular cardiac mesenchymal tumor contained spindle cells, chondroid, osteoid, and abundant collagenous and elastic extracellular matrix (Figure 3b–d). Transmission EM was performed on atrial masses (Figure 3e). Ultra-thin sections confirmed the presence of abundant extracellular matrix, some fibrin, and relatively poorly differentiated spindle-like cells in the tumor mass found in hearts of Vprb TGs (16–20 weeks; Figure 3e).

Light microscopic whole-mount and high-power views (a–d) and ultrastructural features (e) of Vprb TG heart (16–20 weeks). (a) bilateral atrial masses are visible on H&E whole-mount histology in Vprb TG. The right ventricle and left ventricle (RV, LV) are dilated and show wall thickening bilaterally (original mag × 0.5). At higher power, (b–d) histopathological features in the atrium of Vprb TG include mesenchymal cell proliferation with osteoid and chondroid formation (b; Masson stain), fibrous connective tissue (c; Masson stain), and dense elastic tissue (d; Elastica von Giesson; original mag: × 400 each b–d). TEM examination of cells (e) from the atrial tumor of Vprb TG reveals poorly differentiated cells without sarcomeric elements. Cells are embedded in a matrix that includes fibrin (original mag: × 25 600).
Pathological features in Vprb hearts occurred before severe CHF developed (Figure 4a, b). Hearts from 8–12-week-old Vprb TGs exhibited small atrial subendocardial cellular masses that protruded into the atrial cavity and included irregular groups of cells with interspersed connective tissue. The cells within the aggregates most closely resembled cardiac myocytes (with pink cytoplasm containing striations from cardiomyocyte sarcomeres), but noncardiac cells and interstitial cells also were present. Inspection of cardiomyocyte cytologic detail revealed sarcoplasmic disorganization, multinucleation, appearance of defective (or abortive) mitotic spindles, and dispersed fragmented chromatin within the sarcoplasm (Figure 4b). Similar pathological features were present in hearts from other Vpr lines, but occurred at later time points in the natural history (data not shown), suggesting a dose–effect of the TG in the lines.

Light microscopy of cardiomyocytes and atrial structure of Vprb TG heart (12 weeks): (a) whole-mount view reveals atrial mass composed primarily of cardiomyocytes (Masson trichrome; original magnification, × 0.5). Inset, top: box indicates cross-section of the atrial mass with irregular cardiomyocytes (Masson trichrome; original magnification, × 40). Inset, bottom: cytological details of cardiomyocytes with increased density of connective tissue matrix (Masson trichrome; original magnification, × 200). (b) Cytological details of cardiomyocytes from the mass reveals nuclear envelope scalloping and irregular borders (red arrow), abnormal mitoses, and fragmented nuclear debris in the cytoplasm (yellow arrows; H&E original magnification, × 400).
Nuclear morphology also was disturbed in the atrial cardiomyocytes within these cellular masses. Nuclear envelopes from cardiomyocytes were irregular, scalloped, and contained sharp edges (Figure 4b). Cells outside the masses demonstrated nuclei that were more characteristic of normal cardiomyocytes, but atrophied and hypertrophied myocytes also were found.
Pathological Features of Vpr and Nef TG Hearts
For Vpu and Nef TGs, there were no discernable changes in mortality or fertility, no gross or microscopic changes in myocardial structure or function, and no ultrastructural features to suggest cardiac dysfunction (data not shown).
Cardiac MRI of TG Hearts
Cardiac MRI was performed on Vprb TGs and WT littermates to determine the pathophysiological effects of Vpr on the TG. A 4-chamber view was obtained on Vprb TGs. Data from these studies revealed defective atrial and ventricular contraction and bilateral atrial filling defects in the Vprb heart. The latter finding was consistent with an atrial intracavitary mass (Figure 5).

MRI image of Vprb TG heart. MRI was performed as described. Four-chamber and short axis views for functional analysis were obtained. CINE images were generated at 10 frames per TR with TR set slightly less than the period of the HR. MRI of Vprb TG heart reveals four-chamber cardiac dilation (circle) with right and left atrial filling defect.
ECG Findings from TGs
Vpra,b,h (n=4, 16, 4, respectively) and WT littermates (n=16) were evaluated using real-time ECGs without confounding effects of anesthesia. Electrocardiographic abnormalities were found in TG line Vprb and Vpra,h (Figure 6) when all lines were compared at the same age (12 weeks). Vprb revealed dramatic bradycardia (621±47 bpm) compared to WT littermates (791±8 bpm; Figure 6a; P<0.001). Correlative prolongation of R–R and P–R interval were found in Vprb (90±3 ms) compared to WT (77±2; Figure 6a–c; P<0.001). Interestingly, Vpra exhibited HR similar to that of WT (Figure 6a). No difference in HR variability could be detected (data not shown). The duration of the QRS complex was prolonged to 12±0.3 ms in Vprb TGs, compared with Vpra and WT (10±0.1 ms; P<0.01) cohorts (Figure 6d, e). The Q–T intervals were 44±0.5 for Vprb and 38±0.5 for WT (P<0.001). Importantly, Vprb exhibited the longest QRS complex and Q–T interval of any TG.

(a–e) Quantitative data from real-time ECG taken in unanesthetized WT, Vprb, Vpra, and Vprh male mice: (a) Vprb revealed bradycardia compared to WT littermates (P<0.001). (b, c) Prolongation of R-R and P-R interval were found in Vprb compared to WT (P<0.001). (d, e) Duration of the QRS complex was prolonged in Vprb TGs compared to Vpra,h and WT (P<0.01) and Q–T intervals were similarly increased (P<0.001).
Expression of Cardiac and Connective Tissue Markers in Vpr TG Tissues
The complex pathological and physiological phenotype required a broadly based strategy to unravel its molecular phenotype. To better define subcellular events, and to relate those to the other findings, microarray analyses were performed on RNA extracted from atrial tissue of Vprb TG and WT littermates (12 weeks; n=3 per cohort; Table 1) and were replicated. As shown in Table 1, RNA expression levels were increased in a large number of analytes. Importantly, sample-to-sample individual replicates resulted in data that were reproducible (as recommended by the manufacturer, 3 times per individual sample; not shown). Data were reviewed and quantitative RT-PCR confirmed all of the trends found from the microarray.
Based on the physiological and pathological features of the TG atrium, working hypotheses were formulated that accounted for increased expression of extracellular matrix moieties in a pattern that underscored the pathological phenotype. As may be reasonably expected based on the abundant extracellular matrix in the atrial masses (≥12 weeks), collagen mRNAs increased by an average of 2.4-fold. Consistent with both osteogenesis in the atrial mesenchymal tumors and cardiac decompensation, RNA encoding bone morphogenic protein 4 (bmp-4) was upregulated four-fold and bmp-6 and -7 were upregulated 2–3-fold. Other elements of the extracellular matrix (osteomodulin, fibulin, chondroadherin) also were upregulated (Table 1).
Discussion
The cardiac phenotype that resulted from targeted Vpr expression was robust, gene/dose related, and followed mRNA abundance in the tissue target. Our TG lines (called Vpra,b,c,d,e,h) offered the advantage of targeted Vpr with an organ-specific phenotype without other confounding variables associated with AIDS per se. To define mechanisms of dysfunction throughout the natural history of disease, a variety of pathological, physiological, and molecular approaches clarified aspects of the phenotype. The gene/dose-related TG phenotype in vivo underscores in vitro and in vivo findings that point to Vpr effects on nuclear morphological abnormalities that are considered hallmarks of Vpr damage to terminally differentiated cells.8, 9
The α-MyHC promoter is extensively used,10 and is robust and cardiac-specific. It exhibits excellent tissue specificity and minimal positional effects of insertion.11 Although positional effects of insertion could not be entirely ruled out, phenotypic similarity and gene dose effects among all of the Vpr lines described herein support the specificity in some ways. Although indirectly supportive, findings from TGs with cardiac-targeted Nef and Vpu each independently yielded no cardiac structural or functional phenotype. Taken together with the findings from the Vpr TGs, and our previous findings in which an oxidative stress phenotype was observed in cardiac-targeted HIV Tat,13 the Vpr TG here offers insights into effects of other HIV structural and accessory proteins on cardiac function and structure.
The impact of Vpr on the development of complications in AIDS has been relatively underexplored, particularly in AIDS CM. As HIV Vpr circulates freely in plasma,3 paracrine effects from this retroviral accessory protein could impact cell function (including cardiomyocyte function) in AIDS. Based on data herein, it may be reasonable to suggest that circulating Vpr of sufficient concentration could cause deleterious effects on the cardiomyocyte in analogous ways to observed Vpr effects on immunocytes (defined by others8, 9).
Although the Vpr concentration in cardiac myocytes in TGs may be significant in this model system, the remarkable changes that result in TG hearts and the consistency of the phenotype throughout the TG lines together highlight the potential role of Vpr in alterations of cardiomyocyte cell cycle events. This may be particularly important in AIDS patients in whom the HIV plasma viral load is substantial. Vprb exhibited high expression and robust phenotype early in life whereas Vpra and Vprh had longer survival, slower development of atrial tumors, and later development of signs of heart failure. In all Vpr TG lines, pups developed and matured normally, became fertile adults, reproduced, and bore normal litter sizes that exhibited Mendelian distribution of the hemizygous transgene as may be expected with α-MyHC promoter-driven TGs. Artifacts have been reported with extremely high gene expression of native eukaryotic genes.11
Pathological examination of the Vpr hearts revealed a number of unusual findings. First, the atrial intracavitary masses that developed relatively earlier in the life of Vprb TGs (≈8–12 weeks) were cellular and composed principally of cells with features of cardiomyocytes. In contrast, cytological features of atrial tumors in the older animals of that line (≈16–20 weeks) revealed mesenchymal differentiation more prominently and increased matrix. Cytological findings in cardiomyocytes included an abnormal nuclear envelope, multiple mitotic spindles in myocytes, and intracytoplasmic fragments of chromatin. These findings corroborated in vitro data that showed Vpr abnormal nuclear morphology.8, 9
In this transgenic model, tumors in Vpr TGs were anatomically localized to cardiac atria. There was no predilection for left vs right atrium. Curiously, the cellular and tissue architecture in the left and right ventricle remained relatively intact without evidence of tumor or mitosis, but cellular hypertrophy and ventricular wall thickening were present. To our knowledge, mesenchymal tumors have not been reported in the heart in AIDS,21 yet some implications from this model may be biologically important. Atrial tumors have been described in TG mice with targeted SV40 large T antigen.22 Ultrastructural pathological examination of hearts from Vprb revealed relatively primitive cells in a dense matrix including fibrin.23 Pathologic changes in Nef and Vpu TG lines were absent.
In summary, we successfully utilized the α-MyHC promoter to target expression of HIV Vpr to the cardiac myocyte. Dose-related pathological and physiological effects followed TG expression levels among different Vpr lines. In contrast, cardiac-specific targeting of Vpu or Nef showed no structural or functional phenotype. Vpr TG hearts developed abnormal atrial cell proliferation, cardiac dysfunction, dysrhythmias, atrial mesenchymal tumors, and profound CHF. ECG changes were documented at 12 weeks. Most striking changes were seen in Vprb TGs. Molecular features within the atria included expression of genes involved in early cardiac development and of extracellular matrix elements. Future studies with this Vpr TG may elucidate pathophysiological mechanisms that relate the effects of Vpr-induced alterations in cardiac and noncardiac cells.
References
Matsuda Z, Yu X, Yu QC, et al. A virion-specific inhibitory molecule with therapeutic potential for human immunodeficiency virus type 1. Proc Natl Acad Sci USA 1993;90:3544–3548.
Levy DN, Refaeli Y, MacGregor RR, et al. Serum Vpr regulates productive infection and latency of human immunodeficiency virus type 1. Proc Natl Acad Sci USA 1994;91:10873–10877.
Tungaturthi PK, Sawaya BE, Singh SP, et al. Role of HIV-1 Vpr in AIDS pathogenesis: relevance and implications of intravirion, intracellular and free Vpr. Biomed Pharmacother 2003;57:20–24.
Levy DN, Fernandes LS, Williams WV, et al. Induction of cell differentiation by human immunodeficiency virus 1 vpr. Cell 1993;72:541–550.
Stewart SA, Poon B, Song JY, et al. Human immunodeficiency virus type 1 vpr induces apoptosis through caspase activation. J Virol 2000;74:3105–3111.
Lum JJ, Cohen OJ, Nie Z, et al. Vpr R77Q is associated with long-term nonprogressive HIV infection and impaired induction of apoptosis. J Clin Invest 2003;111:1547–1554.
Reilly JM, Cunnion RE, Anderson DW, et al. Frequency of myocarditis, left ventricular dysfunction and ventricular tachycardia in the acquired immune deficiency syndrome. Am J Cardiol 1988;62:789–793.
Chang F, Re F, Sebastian S, et al. HIV-1 Vpr induces defects in mitosis, cytokinesis, nuclear structure, and centrosomes. Mol Biol Cell 2004;15:1793–1801.
de Noronha CM, Sherman MP, Lin HW, et al. Dynamic disruptions in nuclear envelope architecture and integrity induced by HIV-1 Vpr. Science 2001;294:1105–1108.
Dalloz F, Osinska H, Robbins J . Manipulating the contractile apparatus: genetically defined animal models of cardiovascular disease. J Mol Cell Cardiol 2001;33:9–25.
Robbins J . Remodeling the cardiac sarcomere using transgenesis. Annu Rev Physiol 2000;62:261–287.
Subramaniam A, Jones WK, Gulick J, et al. Tissue-specific regulation of the alpha-myosin heavy chain gene promoter in transgenic mice. J Biol Chem 1991;266:24613–24620.
Raidel SM, Haase C, Jansen NR, et al. Targeted myocardial transgenic expression of HIV Tat causes cardiomyopathy and mitochondrial damage. Am J Physiol Heart Circ Physiol 2002;282:H1672–H1678.
Chu V, Otero JM, Lopez O, et al. Electrocardiographic findings in mdx mice: a cardiac phenotype of Duchenne muscular dystrophy. Muscle Nerve 2002;26:513–519.
Chu V, Otero JM, Lopez O, et al. Method for non-invasively recording electrocardiograms in conscious mice. BMC Physiol 2001;1:6.
Sutliff RL, Haase C, Russ R, et al. Cocaine increases mortality and cardiac mass in a murine transgenic model of acquired immune deficiency syndrome. Lab Invest 2003;83:983–989.
Mitchell GF, Jeron A, Koren G . Measurement of heart rate and Q–T interval in the conscious mouse. Am J Physiol 1998;274:H747–H751.
Gehrmann J, Hammer PE, Maguire CT, et al. Phenotypic screening for heart rate variability in the mouse. Am J Physiol Heart Circ Physiol 2000;279:H733–H740.
Lewis W, Haase CP, Raidel SM, et al. Combined antiretroviral therapy causes cardiomyopathy and elevates plasma lactate in transgenic AIDS mice. Lab Invest 2001;81:1527–1536.
Lewis W, Grupp IL, Grupp G, et al. Cardiac dysfunction occurs in the HIV-1 transgenic mouse treated with zidovudine. Lab Invest 2000;80:187–197.
Lewis W . Pathologic changes in the hearts of patients with AIDS. In: Lipshultz SE (ed). Cardiology in AIDS. Chapman & Hall: New York, 1998, pp 233–254.
Delcarpio JB, Lanson Jr NA, Field LJ, et al. Morphological characterization of cardiomyocytes isolated from a transplantable cardiac tumor derived from transgenic mouse atria (AT-1 cells). Circ Res 1991;69:1591–1600.
Narayanan V . Tuberous sclerosis complex: genetics to pathogenesis. Pediatr Neurol 2003;29:404–409.
Acknowledgements
This work was supported by DHHS NIH R01 HL072707 and HL063666 to WL. Robert Santoianni is thanked for thin sectioning and electron photomicrography of the tumor. Diane Paulaskas is acknowledged for administrative support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lewis, W., Miller, Y., Haase, C. et al. HIV viral protein R causes atrial cardiomyocyte mitosis, mesenchymal tumor, dysrhythmia, and heart failure. Lab Invest 85, 182–192 (2005). https://doi.org/10.1038/labinvest.3700222
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.3700222
Keywords
This article is cited by
-
Impact of HIV-1 Vpr manipulation of the DNA repair enzyme UNG2 on B lymphocyte class switch recombination
Journal of Translational Medicine (2020)
-
HIV-1-Associated Left Ventricular Cardiac Dysfunction in Humanized Mice
Scientific Reports (2020)
-
Prevalence of cardiovascular diseases in HIV-infected outpatients: results from a prospective, multicenter cohort study
Clinical Research in Cardiology (2013)
-
Cardiac-Targeted Transgenic Mutant Mitochondrial Enzymes: mtDNA Defects, Antiretroviral Toxicity and Cardiomyopathy
Cardiovascular Toxicology (2008)
-
Decreased mtDNA, oxidative stress, cardiomyopathy, and death from transgenic cardiac targeted human mutant polymerase γ
Laboratory Investigation (2007)
