
Abstract
Local invasiveness is a characteristic feature of glioblastoma that makes surgical resection nearly impossible and accounts in large part for its poor prognosis. To identify mechanisms underlying glioblastoma invasion and motility, we used Transwell invasion chambers to select for a more potently invasive subpopulation of U87MG human glioblastoma cells. The stable population of tumor cells (U87-C1) obtained through this in vitro selection process were three times more invasive than parental U87MG cells and demonstrated faster monolayer wound healing and enhanced radial motility from cell spheroids. This enhanced invasiveness was associated with an 80% increase in matrix metalloproteinase 2 (MMP-2) activation. No differences in expression levels of pro-MMP-2, membrane-type matrix metalloproteinase I (MT1-MMP), or integrin αvβ3 (mediators of MMP-2 activation) were detected. However, U87-C1 cells exhibited two-fold elevation of tissue inhibitor of metalloproteinases (TIMP)-2 mRNA and protein relative to parental cells. Exogenous addition of comparable levels of purified TIMP-2 to parental U87MG cells increased MMP-2 activation and invasion. Similarly, U87MG cells engineered to overexpress TIMP-2 at the same levels as U87-C1 cells also demonstrated increased MMP-2 activation, indicating that an increase in physiological levels of TIMP-2 can promote MMP-2 activation and invasion in glioblastoma cells. However, exogenous administration or recombinant overexpression of higher amounts of TIMP-2 in U87MG cells resulted in inhibition of MMP-2 activation. These results demonstrate that the complex balance between TIMP-2 and MMP-2 is a critical determinant of glioblastoma invasion, and indicate that increasing TIMP-2 in glioblastoma patients may potentially cause adverse effects, particularly in tumors containing high levels of MT1-MMP and MMP-2.
Similar content being viewed by others


Main
Glioblastoma, the most common malignant brain tumor of adults and among the most lethal of all cancers, is characterized by aggressive tumor cell invasion into the surrounding brain, making complete surgical resection nearly impossible. Despite aggressive surgical excision, radiotherapy, and chemotherapy, survival is only modestly prolonged and glioblastoma patients have a median survival of approximately 12 months from the time of initial diagnosis. The identification of mechanisms involved in glioblastoma invasion, particularly those that can be safely and pharmacologically targeted in patients, is a high priority.
Similar to tumor invasion and metastasis in other types of cancer, glioblastoma invasion is believed to involve a complex series of integrated events consisting of tumor cell adhesion, extracellular matrix (ECM) proteolysis, and cell migration within the surrounding microenvironment.1 Among the proteases contributing to ECM turnover, the family of matrix metalloproteinases (MMPs) plays a significant role in tumor invasion and metastasis.2, 3 Synthesized as either secreted or membrane-bound latent enzymes, MMPs require proteolytic activation involving cleavage of a propeptide domain to exhibit enzymatic activity.4
Considerable evidence implicates MMPs, especially matrix metalloproteinase-2 (MMP-2), MMP-9, and membrane type-I metalloproteinase 1 (MT1-MMP) in glioblastoma invasion.5 MMP-2 expression and activation in particular is upregulated in glioblastoma and is associated with its rapid progression to increasing levels of malignancy.6, 7 Cell-mediated pro-MMP-2 activation involves cleavage by MT1-MMP on cell membranes.8, 9 The activity of these MMPs is regulated by the endogenous specific inhibitor tissue inhibitor of metalloproteinases (TIMP)-2, which in excess can bind cell surface MT1-MMP and both latent and active MMP-2 in an inhibitory manner.10, 11 However, TIMP-2 is also essential at lower concentrations for the efficient activation of MMP-2,12, 13 acting as a bridge between MT1-MMP and MMP-2 on the cell surface14 to allow a second TIMP-free MT1-MMP molecule to cleave the propeptide domain of pro-MMP-2.15 Owing to the dual nature of TIMP-2 in regulating MMP-2 activation, in which both a deficiency and an excess of TIMP-2 can prevent MMP-2 activation, small shifts in the balance between TIMP and MMP levels may greatly modulate the invasive phenotype in tumor cells. Further elucidation of these MMP regulatory mechanisms and their roles during tumor progression is critical especially in view of the disappointing results of recent clinical trials with MMP inhibitors.16, 17
The goal of this study was to identify specific mechanisms contributing to glioblastoma invasion by isolating a subpopulation of more invasive cells from the glioblastoma cell line U87MG and comparing their phenotypes for relevant differences. Here, we demonstrate that endogenous up-regulation of TIMP-2 in glioblastoma cells can lead to increased MMP-2 activation and subsequent invasion.
Materials and methods
Reagents and Antibodies
Human collagen type I was from Becton Dickinson (Bedford, MA, USA). Saponin, FITC-phalloidin, and concanavalin A were from Sigma (St Louis, MO, USA). Purified MMP-2 was from Calbiochem (San Diego, CA, USA). Purified recombinant TIMP-2 was kindly provided by H Lu (Amgen, Thousand Oaks, CA, USA). Polyclonal antibodies against the hinge region of MT1-MMP and monoclonal antibodies specific for integrin αvβ3 (clone LM609) were from Chemicon (Temecula, CA, USA). Monoclonal TIMP-2 antibodies (clone 67-4H11) were from Oncogene Research Products (San Diego, CA, USA). β-Tubulin monoclonal antibodies were from NeoMarkers (Fremont, CA, USA). Horseradish peroxidase-conjugated goat anti-mouse or anti-rabbit secondary antibodies for immunoblots were from New England Biolabs (Beverly, MA, USA), and FITC-conjugated goat anti-mouse or anti-rabbit secondary antibodies used in flow cytometry were from Pierce (Rockford, IL, USA).
Cell Culture
The human glioblastoma cell line U87MG was purchased from the American Tissue Culture Collection and was routinely maintained in modified Eagle's medium (MEM) containing 10% fetal bovine serum (Omega Scientific, Tarzana, CA, USA), 1 × penicillin-streptomycin–glutamine solution (Gibco-Invitrogen, Grand Island, NY, USA), 1 × non essential amino acids (Mediatech, Herndon, VA, USA), 1 mM sodium pyruvate, and 0.15% sodium bicarbonate. For media conditioning experiments, cells were seeded in the above medium and allowed to adhere overnight, at which point the medium was changed to serum-free MEM.
Transwell Invasion Assay and Selection of U87-C1 Cells
Six- or 12-well Transwell polycarbonate membrane inserts with 8.0-μm pores (Costar, Cambridge, MA, USA) were coated from the bottom with 10 μg/ml collagen type I. After adding serum-free MEM to the lower compartments, single-cell suspensions in serum-free MEM/BSA (5 mg/ml) were then seeded onto the filters (470 000 cells for six-well, 100 000 cells for 12-well) and incubated for 5 h at 37°C, 5% CO2. Filters were then washed, and cells on the upper surface were removed with cotton swabs. Cells that had invaded and adhered to the lower surface were fixed in 4% paraformaldehyde for 10 min and stained with 0.25% crystal violet. Eight random fields were counted to determine the number of cells invaded. To generate U87-C1 cells, the first 5% of invading cells from U87MG were selected using the same collagen-coated Transwell filters under serum-free conditions. The incubation time required for roughly 5% of cells to invade was empirically determined to be about 4 h, and invaded cells attached to the undersides of the filters were aseptically harvested by brief, gentle trypsinization and cultured in new dishes. To obtain U87-Res cells, noninvading cells on the upper surfaces of the filters were harvested in a similar fashion and cultured.
Wound Assay
As described previously,18 confluent monolayers were scratched with a plastic pipet tip to create a uniform, cell-free wound area that was then inspected at regular time intervals. At each time point eight photographs of each wound were taken and the distance between the opposing edges was measured at two points on each photograph. The distance migrated in micrometers was calculated as the difference of the scratch width at the beginning of the assay and that at each indicated time point.
Spheroid Assay
Tumor cell spheroids were generated by seeding single-cell suspensions at low densities over a layer of 3% agar. Cells were grown for 10–12 days until spheroids formed, at which point spheroids of similar diameter (300 μm) were selected and placed in an uncoated or collagen type I (10 μg/ml)-coated 96-well dish containing serum-free media. Photographs of the spheroids were taken, and after overnight incubation the radial migration distance of each sample was examined and compared.
Actin Immunofluorescence
Cells were grown to ∼75% confluence on uncoated or collagen type I coated (10 μg/ml)-glass coverslips in a 12-well plate. Coverslips were fixed with 2% paraformaldehyde, quenched with 50 mM ammonium chloride, permeabilized with 0.075% saponin, and finally stained with FITC-conjugated phalloidin (Sigma). Images were acquired with an AxioCam digital camera (Zeiss, Germany) mounted on an Axioskop 2 plus microscope (Zeiss) and analyzed using AxioVision software.
Zymography
Gelatinolytic activity of conditioned media was detected by gelatin zymography. Serum-free media conditioned for 48 h (10 μl) were subjected to SDS-PAGE using 8% acrylamide gels containing 0.1% gelatin. The gels were then incubated for 30 min at room temperature in 2.5% Triton X-100 twice, before being transferred to activation buffer (50 mM Tris-Cl pH 8.0, 10 mM CaCl2) for overnight incubation at 37°C. Gels were then stained with Coomassie Brilliant Blue R-250 and briefly destained in 10% acetic acid and 40% methanol. Gelatinolytic activity was detected as transparent bands on a blue background. Purified human MMP-2 was run alongside conditioned media to confirm the identity of MMP-2 (not shown). Reverse zymography was performed the same way, except that samples of media (30 μl) were run in 15% acrylamide gels containing 0.225% gelatin and 50 ng/ml purified MMP-2. TIMP activity was detected as positive staining bands over a clear background.
Immunoblotting
Confluent cells were homogenized in ice-cold lysis buffer (50 mM HEPES, pH 7.0, 150 mM NaCl, 10 mM EDTA, 1.5 mM MgCl2, 1% Triton X-100, 1% Nonidet P-40, 0.5% sodium deoxycholate, 1 mM phenylmethylsulfonyl fluoride, 5 μg/ml aprotinin, 5 μg/ml leupeptin, and 1 mM sodium orthovanadate), passed through an 18 gauge needle five times each, and then centrifuged at 14 000 rpm in a microfuge at 4°C for 15 min to pellet cell debris. Protein concentrations of the resulting supernatants were determined using a BCA protein assay (Pierce). Equal amounts of protein (20 μg) in Laemmli sample buffer were loaded on 10% polyacrylamide gels and SDS-PAGE was performed. After transfer to nitrocellulose membranes (Hybond, Amersham Pharmacia, Piscataway, NJ, USA), samples were blocked in TBST with 5% nonfat dried milk and incubated with primary antibodies at 4°C overnight. After washing, blots were incubated with the appropriate HRP-conjugated secondary antibodies at a dilution of 1:5000 for 1 h at room temperature. After washing, bands were visualized by incubating with ECL (Amersham Pharmacia), followed by exposure to Kodak BioMax film (Eastman Kodak). All densitometric analysis was carried out using AlphaEase software version 5.04 (Alpha Innotech, San Leandro, CA, USA).
Semiquantitative RT-PCR
Total RNA was isolated using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). A measure of 2 μg of total RNA was reverse transcribed using the SuperScript First-Strand Synthesis System (Invitrogen), after which 2 μl of first-strand cDNA was amplified in a 50 μl PCR reaction volume using the PCRx Enhancer System (Invitrogen) with the TIMP-2 specific primers 5′-GCC CCC GCC CGC CCA GCC CCC C-3′ and 5′-CGA GAC CCC ACA CAC TGC CGA GGA-3′ (332 bp product). Alternatively, primers for glyceraldehyde-3-phosphate (GAPDH) were used to normalize amplification between samples (5′-GTG AAG GTC GGA GTC AAC GG-3′ and 5′-TGA TGA CAA GCT TCC CGT TCT C-3′, 198 bp product). PCR reactions were initially denatured at 95°C for 2 min, followed by 20, 25, 30, or 35 amplification cycles with denaturation at 95°C for 30 s, annealing at 60°C for 30 sec, and extension at 72°C for 30 s. A 5 μl volume of each reaction was then loaded onto 1.5% agarose gels, stained with ethidium bromide, and the band intensities were analyzed by densitometry.
Flow Cytometry
U87MG and U87-C1 cells were incubated in serum-free media and, where indicated, U87MG cells were treated with 350 or 500 ng/ml exogenous purified TIMP-2, or 30 μg/ml of concanavalin A. The next day, cells were harvested with PBS containing 2 mM EDTA (without Ca2+ and Mg2+), pelleted, and rinsed. Cells were resuspended in PBS with 1% BSA, 0.02% sodium azide and incubated with primary antibodies for 1 h on ice. Following washing, cells were incubated for 30 min with the appropriate FITC-conjugated secondary antibodies at 4°C in the dark, washed, and finally resuspended in the same buffer supplemented with 3 μg/ml of propidium iodide. Cells were sampled with a FACScan flow cytometer (Becton Dickinson, Mountain View, CA, USA) using Cell Quest acquisition software (Becton Dickinson) and FCS Express Version 2 analysis software (De Novo Software). Data are presented as mean±s.e.m. of log fluorescence intensity determined in three independent experiments. Significance of difference was determined using the independent t-test.
TIMP-2 Plasmid Construct and Transfection
The complete human TIMP-2 cDNA was generated from U87MG total RNA (500 ng) by RT-PCR as described above, except that PCR was performed using Platinum Taq High Fidelity (Invitrogen) with the full-length TIMP-2 specific primers 5′-GCC CCC GCC CGC CCA GCC CCC C-3′ and 5′-AGG CCT GCT TAT GGG TCC TCG ATG-3′. The amplified cDNA was cloned into pCR2.1-TOPO using the TOPO TA cloning kit (Invitrogen), and the EcoRI excised TIMP-2 fragment was subsequently subcloned into the pcDNA3.1 expression vector (Invitrogen). Orientation and sequence of the TIMP-2 construct were verified by DNA sequencing. Stable transfections of U87MG cells with the pcDNA3.1-TIMP-2 plasmid were carried out using FuGENE 6 (Roche, Indianapolis, IN, USA) as recommended by the manufacturer. Individual colonies of cells resistant to 0.5 mg/ml G418 sulfate (Mediatech) after 2 weeks of selection were isolated with cloning rings and expanded. The TIMP-2 expression level of each clone was determined by reverse zymography of conditioned media. U87MG cells transfected with empty pcDNA3.1 vector and selected in parallel served as controls.
Results
Selection of U87-C1, a More Invasive Subpopulation of U87MG Glioblastoma Cells
To establish a subpopulation of more invasive glioblastoma cells from the U87MG cell line, parental U87MG cells were seeded onto Transwell filters coated with collagen type I and incubated for 4 h, the empirically determined time allowing approximately 5% of the total number of cells to invade. We chose to use collagen type I as a matrix barrier because it has been shown to be ectopically upregulated in glioblastoma tissue.19 Cells selected on other ECM proteins such as vitronectin and collagen type IV demonstrated similar phenotypes and were not studied further. The selection process was performed under serum-free conditions in the absence of chemoattractants so that only differences in cell behavior inherent to the tumor cells would be isolated, and not those due to external cues. Because cells that had invaded through the Transwell membrane adhered to the underside of the filters and did not progress to attach to the wells of the lower chamber compartments, the invasive cells were harvested by brief and gentle trypsinization from the bottoms of the filters and cultured in new dishes. The selected cells were subsequently expanded and designated as U87-C1. Residual cells that had not invaded and remained on the upper surface of the filters were harvested for comparison and designated as U87-Res. The cellular and molecular phenotypes of the selected cells, described below, were stable at least up to 15 passages in our experiments. There were no clearly discernable differences in cell morphology between the parental U87MG cells, U87MG-Res, and U87-C1 cells under phase-contrast microscopy.
U87-C1 Cells Exhibit Increased Invasiveness and Motility
Differences in invasive and motile behaviors associated with selected U87-C1 cells compared to parental cells were characterized using three independent methods. Interestingly, a single round of selection at this level of stringency sufficiently isolated a stable subpopulation of glioblastoma cells with enhanced invasion. Transwell chambers coated with collagen type I clearly showed that U87-C1 cells exhibited a marked increase in invasion over parental cells (Figure 1). The invasive capacity of the selected cells was consistently increased three-fold in four independent assays carried out in triplicate (P<0.0001). The invasion rate of residual U87-Res cells was nearly identical to that of parental cells. To confirm this phenotype independently, differences in motility were assessed using a wound assay in which cells are grown to confluence in monolayers, and their ability to migrate into and across a linear cell-free area of the monolayer is followed over time. U87-C1 cells readily migrated into the wound within 2 h (Figure 2a) and were able to close the wound within 8 h, whereas parental cells migrated much slower in a more uniform, less diffuse and infiltrative pattern. The migration of U87-Res cells was diminished by about 30% compared to parental cells. No differences in cell proliferation between U87MG and U87-C1 cells were detected using BrdU incorporation and MTS assays (data not shown), ruling out the possibility that the selected cells appeared more motile due to increased cell division. As an additional independent confirmation, differences in motility were also analyzed using a spheroid radial migration assay. Tumor cell spheroids grown on nonadherent surfaces to approximately 300 μm in diameter were individually seeded onto either uncoated or collagen type I-coated wells and allowed to adhere and migrate overnight. Consistent with the Transwell and wound-closing assay results, U87-C1 cells migrated distances two-fold greater than parental cells away from the initial spheroids in a radial pattern (Figure 2b). Selected cells were able to diffusely migrate out a distance of about 300 μm in each direction, leaving little of the original spheroid structure discernable. The migration of cells from parental spheroids was more compact, the resultant spheroids being slightly smaller in diameter but their shape clearly intact. Assays performed on collagen substrates slightly enhanced the radial motility in both cell lines, but selected cells still demonstrated markedly more diffuse migration. These results demonstrate that selected U87-C1 cells display enhanced invasiveness and motility in multiple independent assays.

U87-C1 cells were selected from the parental U87MG cell line as described in ‘Materials and methods.’ U87-C1 cells were subsequently expanded and their invasion through collagen type I (10 μg/ml)-coated Transwell filters was compared to parental cells. (a) Representative photomicrographs of invaded cells on collagen-coated filters. Equal numbers of cells were seeded and incubated for 5 h in serum-free media. Cells that had penetrated onto the lower surface of the filter were fixed in paraformaldehyde and stained with crystal violet. Bar, 100 μm. (b) Quantitation of cell invasion. Eight random fields from each Transwell were photographed and stained cells were visually counted. Invasion of U87-C1 cells was consistently three-fold greater than U87MG, while U87MG and U87-Res cells displayed equivalent levels of invasion. Error bars represent s.e.m. of the mean of four independent assays done in triplicate. Significance of difference between U87MG and U87-C1 invasion was estimated using the independent t-test.

U87-C1 cells are more motile than parental U87MG cells in wound healing and spheroid radial migration assays. (a) Migration of cells was routinely monitored after confluent monolayers were gently scratched with a plastic pipet tip in the wound assay. U87-C1 cells closed the wound by 8 h. U87-Res cells were less motile than both U87-C1 and U87MG cells. Error bars in the chart represent s.e.m. of the mean of 10 independent experiments. *Significance of difference between U87MG and U87-C1 migration at different time points was estimated using the independent t-test and were both P<0.005. Bar, 500 μm. (b) Spheroids approximately 300 μm in diameter were seeded onto BSA or collagen type I-coated surfaces and allowed to adhere and migrate overnight. Representative photographs of the spheroids before and after migration are shown. Bar, 250 μm.
U87-C1 Cells Display Cytoskeletal Features of Motility
To determine whether the differences in invasion and motility correlated with cytoskeletal alterations associated with tumor cell invasion, we performed immunofluorescence staining of the actin cytoskeleton using FITC-phalloidin. U87-C1 cells exhibited extensive lamellipodia (Figure 3, arrows) in addition to the presence of well-defined stress fibers (arrowheads). On the other hand, parental cells presented fewer stress fibers and lacked the lamellipodia indicative of cell motility found in the selected cells. Plated on collagen-coated surfaces, both cell types demonstrated a slight increase in actin-based membrane specializations, but again the differences between U87MG and U87-C1 cells remained. These results indicate that U87-C1 cells feature cytoskeletal rearrangements that correlate with their enhanced invasiveness.

Staining of the actin cytoskeleton with FITC-phalloidin revealed extensive lamellipodia (arrows) as well as formation of organized actin stress fibers (arrowhead) in U87-C1 cells.
U87-C1 Cells Exhibit Increased Levels of proMMP-2 Activation
To elucidate the molecular differences underlying the increased invasiveness and motility of U87-C1 cells, zymographic analysis of conditioned media was performed to assess whether the more invasive phenotype correlated with increases in MMP-2 and MMP-9 expression and/or activation. In all the cell lines, a 72 kDa gelatinase activity corresponding to proMMP-2 was robustly observed at equivalent levels. In conditioned media from U87-C1 cells, however, there was an 80% increase in both 64 kDa intermediate and 62 kDa fully activated MMP-2 processed forms compared to parental and residual cells, which had comparable levels of MMP-2 activation (Figure 4). This increase in MMP-2 activation in U87-C1 cells was observed whether the cells were grown in media with or without serum supplementation (Figure 4a), indicating that the effect was not a result of differential responsiveness to serum-borne factors. Overall pro-MMP-2 expression, as detected by zymography and immunoblot, did not differ between parental and selected cells, indicating a difference specifically in the amount of MMP-2 activation in U87-C1 cells. In contrast, MMP-9 expression and activation were identical in both parental and selected lines (data not shown).

U87-C1 cells activate MMP-2 more efficiently than U87MG and U87-Res. (a) Media conditioned for 48 h under both serum and serum-free conditions were analyzed for MMP-2 activation by gelatin zymography. (b) Densitometric analysis of MMP-2 bands from zymography showed an 80% increase in the amount of 64 kDa intermediate and 62 kDa fully activated MMP-2 species in U87-C1 cells over parental, while MMP-2 activation was comparable between U87MG and U87-Res cells. Activation level is presented as the mean densitometric ratio compared to U87MG over 15 independent samples; error bars represent s.e.m. of this mean. Significance of difference in MMP-2 activation between U87MG and U87-C1 was estimated using the independent t-test.
Increased MMP-2 Activation in U87-C1 Cells is Not Associated with Increased MT1-MMP or Integrin αvβ3 Expression
In the well-characterized model mechanism of cell-mediated MMP-2 activation, MT1-MMP on the cell surface forms a complex with TIMP-2, which serves as a receptor for pro-MMP-2 binding.20 A second TIMP-free MT1-MMP molecule in close proximity to this ternary complex then cleaves the propeptide domain of proMMP-2, forming an activated intermediate that is subsequently processed to the fully activated form through intermolecular autocleavage. Furthermore, integrin αvβ3 has been shown to bind active MMP-2, presenting it on the cell surface to facilitate matrix degradation, as well as to bind proMMP-2 directly for activation by MT1-MMP.21, 22 To determine whether there were any changes in expression of these molecules that could account for increased MMP-2 activation in U87-C1 cells, the expression levels of MT1-MMP, TIMP-2, and integrin αvβ3 were individually analyzed.
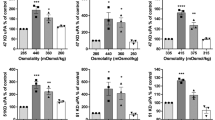
Immunoblot analysis on cell lysates revealed no detectable changes in MT1-MMP levels (Figure 5a). In both cell lines, a 63 kDa band corresponding to latent MT1-MMP and a 55 kDa band representing activated MT1-MMP were present at equivalent levels. A faint 43 kDa band presumably representing the processed inactive form of MT1-MMP was also observed equally in both types of cells. Levels of MT1-MMP cell-surface expression under normal culture conditions were also determined after staining cells with MT1-MMP-specific antibodies followed by FACs analysis. The amounts of MT1-MMP expressed on the cell surface corresponded with the immunoblot data, with identical staining patterns and intensities for both cell lines (Figure 5b).

U87-C1 cells express equivalent levels of MT1-MMP and integrin αvβ3 as parental U87MG cells. (a) Immunoblot analysis of cell lysates (20 μg) using polyclonal antibodies against the hinge region of MT1-MMP following SDS-PAGE on 10% acrylamide gels revealed the same amounts of 63 kDa pro-MT1-MMP, 55 kDa active MT1-MMP, and 43 kDa inactive MT1-MMP between U87-C1 and U87MG. β-Tubulin was detected to ensure equal protein loading. (b and c) Cytofluorimetric analyses of MT1-MMP and integrin αvβ3 using specific antibodies show equivalent cell-surface staining.
Similarly, cell-surface expression of αvβ3 in U87-C1 cells coincided with that of the parental cells as determined by flow cytometry using a monoclonal antibody against the heterodimer (Figure 5c), suggesting that there was no increase in interaction between the integrin and MMP-2 that might facilitate protease activation and activity.
Increased MMP-2 Activation in U87-C1 Cells is Accompanied by Increased TIMP-2 Expression
We next studied expression of soluble TIMP-2 in the conditioned media of the cells by immunoblotting and reverse zymography. Reverse zymography of conditioned media revealed that the level of soluble TIMP-2 protein from U87-C1 cells was increased two-fold over that of parental and U87-Res cells (Figure 6a), as quantitated by densitometry. Likewise, immunoblots on 10x concentrated conditioned media using a monoclonal antibody against TIMP-2 indicated two-fold higher levels of secreted TIMP-2. Gelatin zymography of the same conditioned media samples confirmed that MMP-2 activation in U87-C1 cells was increased in the presence of higher levels of secreted TIMP-2. Semiquantitative RT-PCR was also performed using total RNA to assess whether the increase in TIMP-2 secretion in U87-C1 cells was a result of higher TIMP-2 mRNA levels. Following reverse transcription with oligo(dT) primers, a 332 bp PCR product corresponding to the 5′ end of the TIMP-2 transcript was amplified from both cell lines (Figure 6b). However, after 30 PCR cycles the amount of product generated was 75% greater in U87-C1, suggesting that the difference in TIMP-2 levels occurred at the level of transcriptional regulation.

Selected U87-C1 cells express and secrete increased amounts of TIMP-2 into conditioned media. (a) Media from U87-C1 cells with increased MMP-2 activation showed two-fold higher levels of TIMP-2 by reverse zymography and immunoblot. TIMP-2 expression was the same in both parental U87MG and residual U87-Res cells. Conditioned media were concentrated 10-fold using Centricon YM-10 centrifugal filter columns (Millipore, Bedford, MA, USA) for immunoblots. (b) Semiquantitative RT-PCR was performed on total RNA using primers that amplify a 332 bp 5′ fragment of the TIMP-2 transcript. PCR reactions were amplified for 20, 25, 30, or 35 cycles, and 5 μl aliquots were run on ethidiumbromide-stained 1.5% agarose gels. RT-PCR reactions for U87-C1 demonstrated a 76% increase in amplified TIMP-2 product over parental cells after 30 PCR cycles. Primers specific for GAPDH were also used as a normalization control. Densitometric ratios of band intensities are indicated.
Exogenous TIMP-2 Increases MMP-2 Activation and Cellular Invasion
To determine whether an increase in local TIMP-2 concentration could promote proMMP-2 activation in U87MG cells, purified TIMP-2 was exogenously added to the culture medium of cells at varying concentrations. In U87MG cells, the addition of TIMP-2 stimulated MMP-2 activation in a dose-dependent manner at concentrations ranging between 50 and 700 ng/ml, as seen by zymography (Figure 7a). At concentrations in excess of 900 ng/ml, exogenously added TIMP-2 was inhibitory to proMMP-2 activation. (Actual TIMP-2 concentrations in the media are in reality higher to account for endogenously expressed TIMP-2.) Exogenous addition of TIMP-2 to U87-C1 cells likewise stimulated proMMP-2 activation in a dose-dependent manner, except that slightly higher levels of the activated intermediate and fully processed forms were achieved with lower TIMP-2 doses. A measure of 200 ng/ml of TIMP-2 added to media of U87-C1 cells generated roughly the same maximum amounts of activated MMP-2 products as 700 ng/ml of TIMP-2 added to parental U87MG cells. A decline in MMP-2 activation began to be observed in U87-C1 cells given 300 ng/ml of TIMP-2, whereas the same was seen in U87MG cells at 800 ng/ml of TIMP-2. Exogenous TIMP-2 at a concentration of 1000 ng/ml had an inhibitory effect on MMP-2 activation in both cell lines. These data suggest that endogenous levels of TIMP-2 in U87-C1 cells are roughly 500 ng/ml higher than parental U87MG cells.

Addition of exogenous TIMP-2 increased MMP-2 activation and cell invasion in a dose-dependent manner. (a) Exogenous recombinant human TIMP-2 was added to freshly seeded cells at various concentrations and incubated for 48 h in serum-free media. Aliquots of conditioned media run on gelatin zymography showed that the addition of TIMP-2 up to 700 ng/ml in U87MG cells steadily increased MMP-2 activation, whereas higher concentrations were inhibitory. U87-C1 cells activated MMP-2 increasingly with TIMP-2 concentrations up to 200 ng/ml, while inhibition of protease activation gradually ensued at 300 ng/ml. The shift in exogenous TIMP-2 concentrations in relation to MMP-2 activation indicated that U87-C1 cells secreted about 500 ng/ml more TIMP-2 than parental cells. (b) U87MG cells were more invasive through collagen-coated Transwells when they were incubated with exogenous TIMP-2. Invasion was enhanced by up to 40% in the presence of 300 ng/ml TIMP-2. Data represent mean±s.e.m. of three independent experiments. Significance of difference (P<0.05) was estimated by comparing data between assays with exogenous TIMP-2 and assays without TIMP-2.
The effects of increased MMP-2 activation mediated by TIMP-2 addition on cell invasion were also examined in Transwell assays coated with collagen type I. Exogenous TIMP-2 concentrations of 100 and 300 ng/ml increased cell invasion in a dose-dependent manner by about 20 and 40%, respectively, as seen in Figure 7b. Collectively, these results indicate that a moderate increase in local TIMP-2 concentrations in U87MG cells can promote proMMP-2 activation, thus correlating the higher levels of TIMP-2 expression in U87-C1 cells with their increased MMP-2 activation and cellular invasion.
Since TIMP-2 binding of cell-surface MT1-MMP is essential for subsequent MMP-2 binding and activation, we next assessed whether TIMP-2 levels on the cell surface of U87-C1 cells or on U87MG cells preincubated with exogenous TIMP-2 were increased in accordance with their higher local TIMP-2 concentrations and increased MMP-2 activation. Surface staining of cells with a TIMP-2 specific antibody followed by FACs analysis revealed that surface-bound TIMP-2 was increased by over 30% on U87-C1 cells compared to parental U87MG (Figure 8). Similarly, incubation of U87MG cells with exogenous TIMP-2 resulted in a dose-dependent increase of cell-bound TIMP-2. An amount of 350 ng/ml of TIMP-2 that was added to the media of U87MG cells increased cell-bound TIMP-2 to a level slightly higher than that observed in U87-C1 cells, whereas 500 ng/ml of exogenous TIMP-2 increased TIMP-2 surface binding nearly two-fold. As a control, U87MG cells treated with the lectin concanavalin A, a potent inducer of MMP-2 activation, also exhibited a dramatic increase in cell-bound TIMP-2. These results suggest that the higher expression of TIMP-2 in U87-C1 cells led to an increase in TIMP-2 binding to cell-surface MT1-MMP and ensuing MMP-2 activation.

Increased expression and exogenous addition of TIMP-2 results in higher levels of cell-surface-bound TIMP-2 as determined by FACS. U87-C1 cells showed over a 30% increase in cell-surface TIMP-2 compared to U87MG. The addition of exogenous TIMP-2 to U87MG increased surface-bound TIMP-2 in a dose-dependent manner, suggesting that cell-surface MT1-MMP binding sites were not fully saturated at these TIMP-2 concentrations. U87MG cells treated with concanavalin A to stimulate MMP-2 activation served as a positive control. Data are presented as mean±s.e.m. of log fluorescent intensity. Significance of difference between U87MG and U87-C1 was estimated in the independent t-test for P<0.05.
Transfection of TIMP-2 in U87MG Cells Demonstrates a Biphasic Pattern of MMP-2 Activation
Parental U87MG cells were next transfected with a eukaryotic expression vector containing the human TIMP-2 cDNA or with empty vector to simulate the effects of endogenous TIMP-2 upregulation. Following G418 selection three TIMP-2 transfected stable clones (U87T2, T3, T4) and one control clone were isolated and expanded. As expected, reverse zymography of serum-free conditioned media from the control clone showed the same levels of TIMP-2 as untransfected U87MG cells (Figure 9). U87T2 and T4 transfected clones, however, revealed a dramatic overexpression of TIMP-2 well in excess of the levels found in selected U87-C1 cells. Meanwhile, U87T3 cells exhibited an intermediate upregulation of TIMP-2 expression similar to U87-C1 cells that was higher than both control transfected and parental U87MG cells, but much lower than the levels found in U87T2 and T4 cells. The same results were observed when immunoblots detecting TIMP-2 were performed. Zymographic analysis of the same conditioned media revealed similar levels of MMP-2 activation in control and untransfected U87MG cells, whereas MMP-2 activation was inhibited in TIMP-2 overexpressing U87T2 and T4 cells. In contrast, a marked increase in MMP-2 activation was seen in U87T3 cells intermediately overexpressing TIMP-2, a phenotype mimicking that of selected U87-C1 cells (Figure 9). These data show that expression of TIMP-2 at levels similar to those found in U87-C1 promotes MMP-2 activation, whereas exceeding levels of TIMP-2 expression inhibit MMP-2 activation in U87MG glioblastoma cells.

Transfection of TIMP-2 cDNA into U87MG cells resulted in several clones with different levels of TIMP-2 expression. Conditioned media from cells were analyzed for TIMP-2 expression by reverse zymography and immunoblot, while MMP-2 activation was observed with gelatin zymography. U87MG cells transfected with empty vector exhibited the same amounts of TIMP-2 expression and MMP-2 activation as parental cells. Three TIMP-2 transfected clones were isolated: clones T2 and T4 highly overexpressed TIMP-2 into media and showed diminished MMP-2 activation, whereas clone T3 demonstrated intermediate upregulation of TIMP-2 with increased MMP-2 activation similar to selected U87-C1 cells.
Discussion
One of the critical challenges in glioblastoma research is the identification of mechanisms used by glioblastoma cells to invade the brain. With a variety of new pathway inhibitors and MMP inhibitors being recently introduced into clinical trials, the importance of discovering mechanisms of invasion that can be pharmacologically targeted is apparent. In this study, we have successfully used an in vitro selection method to generate an interesting and useful experimental model for identifying candidate mechanisms involved in glioblastoma invasion without having to genetically modify cells. Relative to parental U87MG cells and U87MG-Res cells, U87-C1 cells derived from this selection process demonstrated a three-fold increase in invasion, displayed cytoskeletal features characteristic of cell motility, and exhibited biochemical alterations that were stable over at least 15 passages. We show that a two-fold increase in TIMP-2 expression in U87-C1 cells is associated with increased MMP-2 activation and invasion. These observations were confirmed and extended using exogenous TIMP-2 addition and TIMP-2 overexpression at varying levels, demonstrating the dual roles that TIMP-2 plays in regulating MMP-2 processing and invasion. Although previous studies have indicated that TIMP-2 can inhibit MMP-2 activation and that it is also required in this process, this is, to the best of our knowledge, the first demonstration that endogenous physiological upregulation of TIMP-2 expression can promote MMP-2 activation and subsequent glioblastoma cell invasion. Furthermore, because higher expression of TIMP-2 (and MMP-2) in glioblastoma tissue has been previously documented,23 these data have direct clinical relevance.
Elevated MMP-2 expression and activation are strongly correlated with astrocytic tumor grade and malignancy.6, 7, 24 Our finding that the more invasive U87-C1 cells have a three-fold increase in MMP-2 activation is consistent with these reports. Moreover, that increased amounts of TIMP-2 can promote MMP-2 activation and glioblastoma cell invasion is also not surprising. Although TIMP-2 was originally characterized as a suppressor of tumor invasion due to its ability to bind and inhibit MMP-2,11 and overexpression of TIMP-2 was previously shown to reduce invasion and metastasis in a number of tumor cell models,25, 26, 27 it is well known that TIMP-2 is also necessary for the efficient activation of proMMP-2. According to the model, a half molar ratio of TIMP-2 to MT1-MMP is theoretically optimal for MMP-2 activation. Exogenous addition of TIMP-2 to U87MG cells at concentrations up to 700 ng/ml not only stimulated MMP-2 activation in a dose-dependent manner, it also increased cell invasion through Transwell filters coated with collagen type I. Only when TIMP-2 levels were in excess of 800 ng/ml was MMP-2 activation inhibited. Similarly, intermediate upregulation of TIMP-2 in U87MG cells by transfection elicited a marked increase in MMP-2 activation, whereas highly overexpressing TIMP-2 transfectants displayed inhibition of MMP-2 activation. It appears then that endogenous TIMP-2 concentrations in U87MG cells, while enough to provide a basal level of MMP-2 activation, are suboptimal for maximum MMP-2 activation in the context of endogenous expression levels of MT1-MMP and MMP-2 in these cells. Thus, increasing local TIMP-2 concentrations has the effect of initially stimulating MMP-2 activation, whereupon exceeding amounts of TIMP-2 are inhibitory to MMP-2 activation (Figure 10). This inhibition is caused by excessive binding of TIMP-2 to MT1-MMP, reducing the number of TIMP-free MT1-MMP molecules available for proMMP-2 cleavage, as well as direct binding of TIMP-2 to proMMP-2, thus sequestering it from the activation complex.

Diagrammatic model depicting the balance between MMP-2/MT1-MMP and TIMP-2 expression and its effects on MMP-2 activation in glioblastoma. (a) Tumor cells such as U87MG that overexpress large amounts of MMP-2 and MT1-MMP but secrete relatively low levels of TIMP-2 are able to activate MMP-2 at a basal level. TIMP-2 (black) binds to MT1-MMP (dark gray) on the cell surface and acts as a receptor for proMMP-2 (light gray). A second TIMP-free MT1-MMP molecule in close proximity then cleaves the MMP-2 propeptide domain to generate active MMP-2 (white), which is then released. (b) Intermediate upregulation of TIMP-2 expression, such as in U87-C1 cells, allows more MT1-MMP/TIMP-2/proMMP-2 complexes to assemble on the cell surface and results in increased MMP-2 activation. (c) Very high levels of TIMP-2 expression are inhibitory to both MMP activity and MMP-2 activation due to excessive TIMP-2 binding of MT1-MMP as well as direct binding to MMP-2. (d) Relationship between MMP expression, local TIMP-2 levels, and MMP-2 activation. Solid black arrowhead lines represent MMP-2 activation in tumor cells with high MMP-2 and MT1-MMP expression as a function of TIMP-2 level. MMP-2 activation initially increases as TIMP-2 increases until TIMP-2 levels reach the optimum for maximal MMP-2 activation. Thereafter, increases in TIMP-2 are inhibitory to activation and, at higher levels, inhibitory to MMP activity. Points A, B, and C are representative positions along this plot for the scenarios depicted in Figures 10(a)–(c). In nontumor tissues that presumably express low levels of MMP-2 and MT1-MMP, even low amounts of TIMP-2 are sufficient to inhibit MMP activation and activity (gray arrowhead line, bottom left).
Because TIMP-2 has been shown to promote cell proliferation,28, 29 an increase in cell growth resulting from TIMP-2 upregulation could potentially be misinterpreted as increased migratory and invasive phenotypes in our assays. Under our culture conditions, however, this was not the case as U87-C1 cells exhibited no growth advantage over U87MG cells as determined by cell growth and DNA synthesis assays. This observation, together with flow cytometry data showing increased cell-surface binding of TIMP-2 in U87-C1, suggests that in our cells the predominant role of TIMP-2 is as a regulator of MMP activation and inhibition.
The mechanism resulting in increased TIMP-2 expression in U87-C1 cells is not clear at this point. TIMP-2 expression appears to be modulated by a wide range of physiological conditions, including upregulation in the presence of lipopolysaccharides and retinoic acid,30 downregulation by TGF-2β,31 and even coordinate regulation with MT1-MMP expression and proteolytic processing.32 However, the fact that U87-C1 cells were derived in the absence of any external stimuli tends to support an intrinsic mode of TIMP-2 regulation that is likely influenced by other inherent cellular and molecular alterations in the cells.
Although MMPs and their inhibitors play key roles in the dynamic processes of glioblastoma cell invasion and motility, there are likely to be other mechanisms promoting these phenotypes including those not involving proteases. Activation of the small GTPase Rho induces stress fiber formation while Rac contributes to the formation of lamellipodia.33 Our data from actin immunofluorescent staining thus strongly suggest an increase in activation of Rho and Rac in U87-C1 cells. Furthermore, inhibition of MMP activity with the broad-spectrum MMP inhibitor GM6001 in both U87MG and U87-C1 cells diminished overall invasion of both cell types, but did not reduce U87-C1 invasion back to parental baseline levels (data not shown). It is likely that other pathways or intrinsic adaptations exist in U87-C1 cells to trigger an invasive phenotype. In glioblastoma, it is probably a combination of various mechanisms in addition to interactions with the surrounding stromal environment that contribute to this pattern of growth and dissemination within the brain.
In the light of our results, as well as those that have reported poor prognostic significance for increased TIMP-2 expression in other human cancers,34, 35, 36, 37 the outcomes of potential TIMP-based therapeutic strategies may not be as straightforward as the original concept of TIMPs serving as anti-invasion/MMP agents. The development of such therapies would require a careful understanding of the TIMP-MMP balance in each individual patient to better predict their suitability and efficacy. This is made more complicated by the fact that tumor cells may also utilize MMPs and TIMPs produced by stromal cells in the local tumor microenvironment. Because it will be difficult to precisely control the efficiency and exact amount of TIMP-2 delivery to tumors in vivo, regardless of the mode of delivery, it may be possible for TIMP-2 therapy to inadvertently shift some tumor cells closer to the optimum MMP/TIMP balance for MMP-2 activation.
In conclusion, we have selected a subpopulation of glioblastoma cells that exhibit greater invasive and motile characteristics as well as increased MMP-2 activation mediated by an increase in TIMP-2 expression. The effects of local TIMP-2 concentrations on MMP-2 activation are dependent upon their stoichiometric ratios to MMP-2 and MT1-MMP levels. Glioblastoma cells exhibiting MT1-MMP and MMP-2 overexpression but low TIMP-2 levels may be rendered more invasive with appropriate upregulation or recruitment of TIMP-2. Our results conclusively demonstrate the complex roles of TIMP-2 in the regulation of MMP-2 activation and glioblastoma cell invasion, and suggest that a careful analysis of TIMP/MMP balances and other cell-surface molecules in vivo may better identify and refine potential therapeutic strategies targeted against glioblastoma invasion.
References
Stetler-Stevenson WG, Aznavoorian S, Liotta LA . Tumor cell interactions with the extracellular matrix during invasion and metastasis. Annu Rev Cell Biol 1993;9:541–573.
Stetler-Stevenson WG . Dynamics of matrix turnover during pathologic remodeling of the extracellular matrix. Am J Pathol 1996;148:1345–1350.
DeClerck YA . Interactions between tumour cells and stromal cells and proteolytic modification of the extracellular matrix by metalloproteinases in cancer. Eur J Cancer 2000;36:1258–1268.
Nagase H . Activation mechanisms of matrix metalloproteinases. Biol Chem 1997;378:151–160.
Forsyth PA, Wong H, Laing TD, et al. Gelatinase-A (MMP-2), gelatinase-B (MMP-9) and membrane type matrix metalloproteinase-1 (MT1-MMP) are involved in different aspects of the pathophysiology of malignant gliomas. Br J Cancer 1999;79:1828–1835.
Beliveau R, Delbecchi L, Beaulieu E, et al. Expression of matrix metalloproteinases and their inhibitors in human brain tumors. Ann N Y Acad Sci 1999;886:236–239.
Nakada M, Kita D, Futami K, et al. Roles of membrane type 1 matrix metalloproteinase and tissue inhibitor of metalloproteinases 2 in invasion and dissemination of human malignant glioma. J Neurosurg 2001;94:464–473.
Deryugina EI, Bourdon MA, Luo GX, et al. Matrix metalloproteinase-2 activation modulates glioma cell migration. J Cell Sci 1997;110(Part 19): 2473–2482.
Seiki M . Membrane-type matrix metalloproteinases. Apmis 1999;107:137–143.
Itoh Y, Ito A, Iwata K, et al. Plasma membrane-bound tissue inhibitor of metalloproteinases (TIMP)-2 specifically inhibits matrix metalloproteinase 2 (gelatinase A) activated on the cell surface. J Biol Chem 1999;273:24360–24367.
Strongin AY, Marmer BL, Grant GA, et al. Plasma membrane-dependent activation of the 72-kDa type IV collagenase is prevented by complex formation with TIMP-2. J Biol Chem 1993;268:14033–14039.
Caterina JJ, Yamada S, Caterina NC, et al. Inactivating mutation of the mouse tissue inhibitor of metalloproteinases-2 (Timp-2) gene alters proMMP-2 activation. J Biol Chem 2000;275:26416–26422.
Wang Z, Juttermann R, Soloway PD . TIMP-2 is required for efficient activation of proMMP-2 in vivo. J Biol Chem 2000;275:26411–26415.
Strongin AY, Collier I, Bannikov G, et al. Mechanism of cell surface activation of 72-kDa type IV collagenase. Isolation of the activated form of the membrane metalloprotease. J Biol Chem 1995;270:5331–5338.
Itoh Y, Takamura A, Ito N, et al. Homophilic complex formation of MT1-MMP facilitates proMMP-2 activation on the cell surface and promotes tumor cell invasion. EMBO J 2001;20:4782–4793.
Zucker S, Cao J, Chen WT . Critical appraisal of the use of matrix metalloproteinase inhibitors in cancer treatment. Oncogene 2000;19:6642–6650.
Coussens LM, Fingleton B, Matrisian LM . Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science 2002;295:2387–2392.
Rajasekaran SA, Palmer LG, Quan K, et al. Na,K-ATPase beta-subunit is required for epithelial polarization, suppression of invasion, and cell motility. Mol Biol Cell 2001;12:279–295.
Paulus W, Roggendorf W, Schuppan D . Immunohistochemical investigation of collagen subtypes in human glioblastomas. Virchows Arch A Pathol Anat Histopathol 1988;413:325–332.
Zucker S, Drews M, Conner C, et al. Tissue inhibitor of metalloproteinase-2 (TIMP-2) binds to the catalytic domain of the cell surface receptor, membrane type 1-matrix metalloproteinase 1 (MT1-MMP). J Biol Chem 1998;273:1216–1222.
Brooks PC, Stromblad S, Sanders LC, et al. Localization of matrix metalloproteinase MMP-2 to the surface of invasive cells by interaction with integrin alpha v beta 3. Cell 1996;85:683–693.
Deryugina EI, Ratnikov B, Monosov E, et al. MT1-MMP initiates activation of pro-MMP-2 and integrin alphavbeta3 promotes maturation of MMP-2 in breast carcinoma cells. Exp Cell Res 2001;263:209–223.
Lampert K, Machein U, Machein MR, et al. Expression of matrix metalloproteinases and their tissue inhibitors in human brain tumors. Am J Pathol 1998;153:429–437.
Kachra Z, Beaulieu E, Delbecchi L, et al. Expression of matrix metalloproteinases and their inhibitors in human brain tumors. Clin Exp Metastasis 1999;17:555–566.
Rigg AS, Lemoine NR . Adenoviral delivery of TIMP1 or TIMP2 can modify the invasive behavior of pancreatic cancer and can have a significant antitumor effect in vivo. Cancer Gene Ther 2001;8:869–878.
Valente P, Fassina G, Melchiori A, et al. TIMP-2 over-expression reduces invasion and angiogenesis and protects B16F10 melanoma cells from apoptosis. Int J Cancer 1998;75:246–253.
Vergani V, Garofalo A, Bani MR, et al. Inhibition of matrix metalloproteinases by over-expression of tissue inhibitor of metalloproteinase-2 inhibits the growth of experimental hemangiomas. Int J Cancer 2001;91:241–247.
Barasch J, Yang J, Qiao J, et al. Tissue inhibitor of metalloproteinase-2 stimulates mesenchymal growth and regulates epithelial branching during morphogenesis of the rat metanephros. J Clin Invest 1999;103:1299–1307.
Corcoran ML, Stetler-Stevenson WG . Tissue inhibitor of metalloproteinase-2 stimulates fibroblast proliferation via a cAMP-dependent mechanism. J Biol Chem 1995;270:13453–13459.
Braunhut SJ, Moses MA . Retinoids modulate endothelial cell production of matrix-degrading proteases and tissue inhibitors of metalloproteinases (TIMP). J Biol Chem 1994;269:13472–13479.
Stetler-Stevenson WG, Brown PD, Onisto M, et al. Tissue inhibitor of metalloproteinases-2 (TIMP-2) mRNA expression in tumor cell lines and human tumor tissues. J Biol Chem 1990;265:13933–13938.
Annabi B, Pilorget A, Bousquet-Gagnon N, et al. Calmodulin inhibitors trigger the proteolytic processing of membrane type-1 matrix metalloproteinase, but not its shedding in glioblastoma cells. Biochem J 2000;359:325–333.
Schmitz AA, Govek EE, Bottner B, et al. Rho GTPases: signaling, migration, and invasion. Exp Cell Res 2000;261:1–12.
Grignon DJ, Sakr W, Toth M, et al. High levels of tissue inhibitor of metalloproteinase-2 (TIMP-2) expression are associated with poor outcome in invasive bladder cancer. Cancer Res 1996;56:1654–1659.
Murashige M, Miyahara M, Shiraishi N, et al. Enhanced expression of tissue inhibitors of metalloproteinases in human colorectal tumors. Jpn J Clin Oncol 1996;26:303–309.
Ree AH, Florenes VA, Berg JP, et al. High levels of messenger RNAs for tissue inhibitors of metalloproteinases (TIMP-1 and TIMP-2) in primary breast carcinomas are associated with development of distant metastases. Clin Cancer Res 1997;3:1623–1628.
Kallakury BV, Karikehalli S, Haholu A, et al. Increased expression of matrix metalloproteinases 2 and 9 and tissue inhibitors of metalloproteinases 1 and 2 correlate with poor prognostic variables in renal cell carcinoma. Clin Cancer Res 2001;7:3113–3119.
Acknowledgements
Supported by NIH grant NS43147 (PSM) and CA88127 (PSM); an Accelerate Brain Cancer Cure Award to PSM; the Harry Allgauer Foundation through the Doris R Ullmann Fund for Brain Tumor Research Technologies, a Henry E Singleton Brain Tumor Fellowship and a generous donation from the Ziering Family Foundation in memory of Sigi Ziering. KVL was supported by USHHS Institutional National Research Service Award #T32 CA09056.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lu, K., Jong, K., Rajasekaran, A. et al. Upregulation of tissue inhibitor of metalloproteinases (TIMP)-2 promotes matrix metalloproteinase (MMP)-2 activation and cell invasion in a human glioblastoma cell line. Lab Invest 84, 8–20 (2004). https://doi.org/10.1038/labinvest.3700003
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.3700003
Keywords
This article is cited by
-
Glucocorticoid-induced cell-derived matrix modulates transforming growth factor β2 signaling in human trabecular meshwork cells
Scientific Reports (2020)
-
Prognostic values and prospective pathway signaling of MicroRNA-182 in ovarian cancer: a study based on gene expression omnibus (GEO) and bioinformatics analysis
Journal of Ovarian Research (2019)
-
Effects of induction and inhibition of matrix cross-linking on remodeling of the aqueous outflow resistance by ocular trabecular meshwork cells
Scientific Reports (2016)
-
Notch3 Ameliorates Cardiac Fibrosis After Myocardial Infarction by Inhibiting the TGF-β1/Smad3 Pathway
Cardiovascular Toxicology (2016)
-
Cross-talk between NADPH oxidase-PKCα-p38MAPK and NF-κB-MT1MMP in activating proMMP-2 by ET-1 in pulmonary artery smooth muscle cells
Molecular and Cellular Biochemistry (2016)
