
Abstract
The cellular immune response to the human immunodeficiency virus, mediated by T lymphocytes, seems strong but fails to control the infection completely. In most virus infections, T cells either eliminate the virus or suppress it indefinitely as a harmless, persisting infection. But the human immunodeficiency virus undermines this control by infecting key immune cells, thereby impairing the response of both the infected CD4+ T cells and the uninfected CD8+ T cells. The failure of the latter to function efficiently facilitates the escape of virus from immune control and the collapse of the whole immune system.
Similar content being viewed by others
Main
The human immunodeficiency virus (HIV) stimulates strong immune responses by cytotoxic T lymphocytes (CTLs) in infected people1,2, despite causing profound immunodeficiency. In the acute phase of the infection, the CTL response initially follows the rise in HIV in the blood and when that response reaches a peak the virus level falls (Fig. 1); after that there is an inverse relationship between CTL response and virus load3. In the monkey model of HIV infection, CTLs can be depleted in vivo by infusion of antibody specific for the CD8 glycoprotein, which is a characteristic of CTLs. When these animals are infected with simian immunodeficiency virus (SIV), the early control of the virus fails4; if the antibody is instead given during the chronic phase of infection, the virus level rises until the effects of the antibody wear off5. These data imply that CTLs are important in control of the virus, a view supported by in vitro experiments6 and by the frequent selection of virus mutants in vivo that are no longer recognized by CTLs7,8, and that therefore escape immune control.

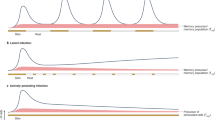
a, An HLA tetramer, based on the known structures of HLA class I (blue), streptavidin (turquoise) and phycoerythrin (red). This can be used to stain antigen-specific CD8+ T cells using fluorescence-activated cell sorting. b, The early CTL response to HIV. The viraemia rose to a peak (red line) and was followed by the CD8+ T-cell response to the immunodominant Gag 263–272 epitope presented by HLA-B27, demonstrated by the HLA-B27–epitope tetramer (mauve line). The viraemia started to fall as the CD8+ T-cell response reached a peak. Further observations were influenced by the initiation of antiretroviral drug therapy (blue bands), but the eventual level of CD8+ T cells specific for a single dominant epitope is typical. (Measurements from ref. 14 in collaboration with C. Workman, Sydney.)
The central role of CTLs in controlling the virus is also emphasized by the influence of human leukocyte antigen (HLA) type on the rate of progress on HIV infection towards AIDS9,10. CTLs recognize virus peptides presented by HLA class I molecules, and different HLA types present different peptides and thus affect the quality of the immune response. HLA types associated with slow progression of the infection, such as HLA-B27 and HLA-B57 (ref. 10), could stimulate more effective immune responses compared with those that confer increased susceptibility, such as HLA-B35 (ref. 9). Similarly, homozygosity at the HLA class I loci, which is also associated with more rapid progression of HIV, offers less opportunity for a diverse T-cell response9.
Although these findings indicate that the CTL response goes some way towards controlling HIV infection, they raise the question of why this response fails ultimately to deal with this virus. This is not the case for other common, persisting human virus infections, such as Epstein–Barr virus (EBV) and cytomegalovirus (CMV)11. So why is HIV different?
CD8 + T-cell response in acute HIV infection
Early analyses of the cellular immune response to HIV in acute infection were measured by a limiting dilution technique where T lymphocytes that responded to HIV were cloned and counted; this showed a strong CTL response coinciding with the initial peak of viraemia12,13 and preceding production of any neutralizing antibody. Better quantification of the early T-cell response can be made with HLA tetramers, where four purified HLA molecules folded around an antigenic virus-derived peptide (epitope) are linked to streptavidin (Fig. 1). Such multimeric reagents bind with sufficient avidity to T-cell receptors to make it possible to identify and count antigen-specific T cells. Using tetramers presenting an immunodominant peptide epitope, the number of HIV-specific T cells was shown to peak just after level of viraemia starts to fall14, suggesting that CTLs are responsible (Fig. 1). A similar pattern is seen in SIV-infected macaques4.
The acute T-cell response is large: as many as 10% of the T cells that carry the CD8 surface protein (which are about half of all T lymphocytes) are involved (refs 14, 15, and C. Spina and P. Hansasuta, unpublished data). Given that the frequency of naive T cells specific for any single HIV epitope must be less than one in a million, such a large reaction involves an expansion in cell numbers requiring at least 17 cell divisions over 2–3 weeks, assuming all progeny survive. In other virus infections, the acute CD8+ T-cell response can be even larger16. Little is known about the function of these early HIV-specific T cells. EBV-specific CD8+ T cells responding to acute infection release cytokines that kill target cells, but they also die by apoptosis or programmed cell death17, which means that their number declines rapidly if antigen does not persist18. Antigen-activated cell death probably controls CTL numbers, but there is a danger that the whole T-cell response might become terminally differentiated and be deleted. This has been seen in mice infected with aggressive lymphocyte choriomeningitis virus (LCMV)19 and may occur occasionally in HIV infection20. The CD8+ T cells responding in acute HIV infection are restricted to a few clones20. Similar restrictions are seen in acute EBV infection (glandular fever)16, where a few dominant clones persist into long-term memory17. In some acutely infected HIV patients, the responding CTLs are monoclonal20, but this carries a bad prognosis, possibly because it risks over-expansion and exhaustion or because it makes it easier for the virus to escape by mutation of the single epitope21.
CD8 + T-cell response in chronic HIV infection
In chronic HIV infection, the expanded HIV-specific T cells persist at high frequencies; often 1–2% of all circulating CD8+ T cells are specific for a dominant HIV epitope3,22. There are similar numbers in lymph nodes23 (V. Appay, unpublished data; G. Pantaleo, unpublished data), indicating that 109 CD8+ T cells can be specific for a single epitope; similar T-cell responses are made to persistent EBV and CMV. These T cells probably turn over continuously24,25; and like the acutely expanded T cells, they tend to die by apoptosis ex vivo26. The high number of responding T cells is almost certainly dependent on continued antigen stimulation, because reduction of HIV level by combinations of potent antiretroviral drugs causes a steady decline in tetramer-stained CD8+ T cells27,28,29. Without treatment, the high number of HIV-specific CD8+ T cells often persists into late infection and they can still be detectable when AIDS develops.
As in acute infection, the expanded T cells are often oligoclonal30, distorting the distribution of T cells with different families of T-cell receptor in the blood. Persistence of T-cell clones marked by their receptors for over five years indicates that either HIV-specific CD8+ T cells are long lived31,32 or they are hard to exhaust if they are continuously in cycle24.
Dynamics of the CD8 + T-cell response
The huge number of CD8+ T cells observed in acute virus infections33,34 implies massive initial expansion of virus-specific CD8+ T cells, which is maintained if virus persists but declines rapidly if virus is cleared15. There is a discrepancy between the number of CD8+ T cells detected by tetramers and the number seen by the more traditional limiting dilution assay (LDA)3,22,35,36,37. The LDA requires that the cloned T cells divide at least 12 times in vitro to become detectable in a functional analysis, such as lysis of HIV-infected target cells. Many or most of the tetramer-stained T cells die rapidly ex vivo, and in most38,39 but not all40 studies, only about 10% can be cloned. Thus antigen-stimulated CD8+ T cells, even within the same clone, can be divided into two types — terminally differentiated cells (in large numbers but likely to die) and long-term memory cells (in low numbers but able to grow). There may well be a continuum of T cells in between these extremes, in various states of differentiation (Fig. 2).

Rare antigen-specific naive CD8+ T cells (blue) are stimulated to divide rapidly to generate T cells that can be detected with epitope-specific HLA–peptide tetramers (turquoise and green). The frequency of effector cells (turquoise) is much higher than those that can divide in vitro (green). Persisting HIV antigen maintains the expanded T cells at a high level, but in the absence of further antigen these cells tend to die by apoptosis (turquoise). The population of T cells that is capable of dividing further (green) maintains long-term memory and is likely to continually generate the expanded effectors (turquoise). The model simplifies the different routes of differentiation and there are almost certainly intermediate phenotypes (see Fig. 3).
Function of HIV-specific CD8 + T cells
Virus-specific CTLs possess a range of antiviral activities, which vary in importance in different infections. These include the ability to kill infected cells and to produce cytokines and chemokines. Perforin is a protein, made by CD8+ T cells, that is present in granules, and together with the granzymes is important in triggering target-cell death. Perforin-knockout mice do not recover from LCMV infection, which implies that lysis of infected cells is crucial for the control of this infection41. Although CD8+ T cells are critical, these knockout mice handle other viruses effectively, such as vaccinia and hepatitis B viruses, implying that cytokine production is more important for control of these pathogens42,43,44.
It is not yet clear which functions of CTLs are most important in controlling HIV. They produce cytokines that can affect viral replication45,46. These include interferon-γ (IFN-γ), which inhibits HIV replication47,48, and tumour-necrosis factor-α (TNF-α), which can upregulate viral replication49,50 through activation of the HIV promoter in the virus 5′ long terminal repeat (LTR). HIV-specific CTLs also produce the CC chemokines MIP-1α, MIP-1β and RANTES51,52, which suppress HIV replication53 by competition for, or downregulation of, CCR5 — the cellular co-receptor (with CD4) for the virus. HIV-specific CTLs secrete these antiviral factors at sites of virus replication, efficiently inhibiting virus replication in vitro6. Inhibition involves lytic mechanisms and release of CC chemokines and a partially characterized secreted factor (CD8+ T-cell antiviral factor, or CAF)54,55. CAF blocks LTR-mediated transcription in infected cells56, thereby shutting off virus production. It may, in addition, facilitate the development of latency, where HIV complementary DNA is integrated into the host-cell genome but remains silent and expresses no messenger RNA and no protein.
Cultured HIV-specific CTLs have been shown to lyse HIV-infected CD4+ T cells in vitro57, despite the ability of the virus Nef protein to reduce expression of class I HLA molecules on their surface58. Susceptibility of infected cells to lysis by CTL clones parallels expression of viral proteins such as Gag p24 within the cell and precedes production of extracellular virus59. This means that lysis is a potent weapon against HIV. Lysis is mediated predominantly by perforin and granzymes60, although a minority of CTLs use the alternative route where Fas ligand expressed on antigen-activated CTLs triggers apoptosis in the target cells that also express Fas61.
Findings from several studies show that virus-specific CTLs taken ex vivo can have functional defects that could undermine their control of virus62,63. Although tetramer staining indicates large numbers of HIV-specific T cells are present in acute or chronic HIV infection, this technique alone makes no measurement of their function. When HIV epitope/HLA class I tetramer-staining was combined with intracellular staining for cytokines and chemokines, it was found that most HIV-specific cells in patients with chronic HIV disease produced IFN-γ, TNF-α and MIP-1β on contact with their cognate antigen ex vivo64. This pattern of cytokine secretion by HIV-specific cells was similar to that of CMV-specific cells from HIV-uninfected donors. However, a striking difference was seen in the level of intracellular perforin. Less than 15% of HIV-specific cells contained perforin, which was reflected in poor ex vivo killing of appropriate target cells, compared with CMV-specific cells from the same donors, 50% of which expressed perforin and killed well64. An earlier study had shown low levels of perforin in CD8+ T cells in the lymph nodes of infected patients65,66.
Thus, HIV-specific T cells may be less efficient killer cells than expected, a feature that might not necessarily have been detected in experiments with cultured CTL clones, cited above, because culture can readily modify or select for function. It is uncertain why HIV-specific cells poorly express perforin: these cells lack expression of the glycoprotein CD28 on their surface, but they retain CD27 (ref. 64). In contrast, CMV-specific cells lose expression of both molecules and this loss is thought to mark out mature effector cells67. So, HIV-specific CD8+ T cells in vivo may be immature67 rather than end-stage effectors as first thought (Fig. 3). Recent findings exploring expression of two other surface glycoproteins — CCR7, a lymphoid-organ homing receptor, and CD45RA, a long isotype of CD45 — support this view: immature CD8+ T memory cells were CCR7+CD45RA+, fully mature CD8+ T cells specific for CMV were CCR7−CD45RA+, and HIV-specific CD8+ T cells were CCR7−CD45RA− (ref. 68). Failure of CD8+ T cells to mature could be a consequence of impaired T-cell help, as suggested by experiments in mice infected with LCMV and lacking helper T cells, where responding T cells were also deficient in function (see below)62, although the nature or strength of the antigenic stimulus could also be relevant. It will be interesting to determine whether different CTL functional phenotypes are associated with different clinical outcomes, and whether such diversity might be linked to the presenting HLA type, particularly those associated with good or bad clinical outcomes9,10.

Surface glycoproteins CD45 (RA and RO isotypes), CD27 and CD28 distinguish stages of CD8+ T-cell maturation. Perforin is expressed only at the final stage. CD45RA reappears as these T cells mature to full effectors. The scheme represents a broad pattern for most HIV- and CMV-specific T cells, although there are some exceptions.
Do HIV-specific CTLs fail for lack of help?
These results question the role of the HIV-specific helper T-cell response, mediated by T cells that express CD4 rather than CD8. The question arises because of the susceptibility of the former to HIV infection. Poor CD4+ T-cell responses to HIV and other recall antigens, measured by proliferation of the T cells to antigen in vitro, were found in HIV-infected patients several years ago69. But a minority of HIV-infected people, who maintain high CD4+ T-cell counts in their blood for many years, actually made good proliferative responses to Gag p24 (ref. 70). Initiation of highly active antiretroviral therapy (HAART) early in infection rescues an anti-Gag p24 CD4+ T-cell proliferative response in most patients70. This result is consistent with the observation that HIV-specific CD4+ T cells, detected by rapid antigen-stimulated release of IFN-γ, were found in early infection but disappeared in untreated patients71. However, Pitcher et al.72 used a different technique to detect CD4+ T cells responding to Gag p24 by IFN-γ secretion in patients at all stages of HIV infection, although the HIV-specific T cells were much less frequent than CMV-specific CD4+ T cells in the same people, so there is agreement that the number of responding T cells is low.
CD4+ T cells that are specific for HIV may be exceptionally susceptible to attack and destruction by HIV. In the periphery, HIV binds to DC-SIGN, a glycoprotein expressed on dendritic cells, without infecting them73. This enables dendritic cells to bring HIV into draining lymph nodes early in primary infection. There the dendritic cells form foci, surrounded by CD4+ T cells activated by antigen processed and presented by dendritic cells in their HLA class II molecules. The proximity of HIV-associated or HIV-infected dendritic cells with activated CD4+ T cells, which are especially susceptible to infection, is a lethal mix for the latter74. HIV-specific T cells are thus attacked by HIV and are likely to be specifically deleted early in infection. There is evidence that this process can be reversed when acutely infected patients are given HAART70,75; this results in preservation of proliferative CD4+ T cells and ultimately better control of HIV on or off drug treatment.
The extent to which the impaired CD4+ T-cell help influences the CD8+ T-cell response is now becoming apparent (Fig. 4). Early in untreated HIV infection, some CD4+ T-cell help may be present and the initial CD8+ T-cell response may be similar to that against any other virus. The problems may come later. T-cell help is known to be important for priming the CD8+ T-cell responses76, for maintaining CD8+ T-cell memory77 and for maturing CD8+ T-cell function62.

CD4+ T cells are important for priming dendritic cells to initiate CD8+ T-cell responses. They help maintain memory T cells and are important in maturation of CD8+ T-cell function. All of these actions are impaired by HIV infection. In addition, HIV can directly infect and impair dendritic-cell function.
Evidence that CD4+ T cells are important in priming CD8+ T cells comes from studies in mice that pinpoint CD40L, expressed on activated CD4+ T cells, as crucial in triggering dendritic cells to produce the cytokine interleukin (IL)-12, which in turn is central in initiating the CD8+ T-cell response76. Some viruses can by-pass this step by activating dendritic cells directly76, but it is not known whether HIV can do this. Failure of T-cell help could disrupt the ability of CD8+ T cells to make new primary immune responses to mutant viruses that have evaded the originally dominant CTL responses (see below).
Helper T cells may be important for the maintenance of CD8+ memory. Survival of infused CD8+ T-cell clones in CMV-infected patients depends on the presence of specific CD4+ T-cell help77; adoptively transferred HIV-specific CTLs survive poorly in HIV-infected patients78,79. These T-cell clones used in adoptive transfer studies may be particularly dependent on IL-2 and other factors provided by helper T cells. In contrast, the long survival of particular CD8+ T-cell clones for many years in patients chronically infected with HIV32, when CD4+ T-cell help is known to be damaged, suggests that naturally activated CD8+ T cells may be able to survive better in the absence of help.
The third role for CD4+ T cells in regulating CD8+ T-cell function has been shown in animal models where absence of CD4+ cells resulted in a CD8+ T-cell response that was numerous in terms of HLA tetramer-stained cells, but lacking in function62. This seems particularly pertinent to HIV infection in view of the findings, discussed above, that CD8+ T cells in HIV infection are not fully mature64.
It is clear that patients with high CD4+ T-cell numbers and detectable helper T cells do better, which is consistent with a more effective T-cell immune response and better immune control of the HIV, demonstrated by a low virus load. Current attempts to rescue the helper T-cell response with drug treatment and vaccine immunotherapy may enhance the CD8+ T-cell response with real benefits75. It should also be recognized that CD4+ T cells could have direct antiviral effects, releasing antiviral cytokines and chemokines and killing infected cells. Activated human T cells express class II HLA, so HIV-infected cells should be targets for CD4+ T cells, although the HIV protein Tat can downregulate HLA class II at the surface of HIV-infected cells80.
The role of helper T cells in CTL survival and function is an area that needs more attention. It is certain that CD4+ T-cell function is impaired early in infection, even if the degree of dysfunction is in dispute. Later, CD4+ T-cell numbers decline drastically. Failure of T-cell help must be central in the pathogenesis of HIV infection and could be the critical feature that ultimately undermines immune control. It will be important to determine when T-cell help is lost in relation to the early events in HIV infection that set the virus load and prognosis.
Escape of HIV from the cellular immune response
CD8+ T cells can act against HIV most effectively by killing infected cells before they generate new virus particles. There is a window of about 12 hours from the onset of virus protein synthesis to the budding of new virus particles. During this period, epitope peptides are presented by HLA class I molecules at the surface of infected cells and can be recognized by CTLs and lysed within five hours57. Unless the killing process eliminates HIV rapidly, it should exert a selective force, giving an advantage to cells infected with viruses that have mutated critical amino acids in the dominant epitopes. These infected cells escape lysis and propagate the mutant virus. Selection of such mutants could be especially favoured when there is impairment of CTL function64, so that virus level is higher than it might otherwise be, with more virus replication and therefore more mutation. The availability of potential escape mutants is the product of virus replication and mutation rate, so the balance between the strength of killing and the level of virus replication should determine how frequently virus escape mutants arise (Fig. 5). It has been argued that the inverse relationship between CTL number (detected by tetramers) and virus load implies the existence of defective killers, because at equilibrium a positive correlation would be expected, such as that found for T cells specific for the non-immunosuppressive human T-cell leukaemia virus type 1 (ref. 81). The observed and expected findings can be resolved if HIV starts to suppress the CTL response above a certain virus threshold. CTLs would still suppress virus load but would be progressively less efficient at doing so as virus levels rise (discussed in ref. 82).

When virus load is high, as in acute infection, there is no selective force until the CTLs appear. When the CTLs are maximally effective there may be little net selection because the virus load has been depressed to a low level. In chronic HIV infection, the CTLs may be suboptimal giving rise to many escape mutants because of high virus turnover.
Selection of mutants by CTLs is probably one of the main features of HIV infection. Longitudinal studies of individual patients, matching dominant CTL responses with changes in amino-acid sequence, have identified clear cases where a single change has abrogated presentation by the class I HLA molecules and these viruses have become dominant in the quasispecies, the swarm of virus variants that arise in vivo (Table 1). A good example was found after an attempt to treat a patient with his own CTL clone, infused in very high numbers83. The clone was specific for a Nef epitope presented by HLA-A3, and virus was selected that had deleted the relevant region of nef. This mutated virus accounted for 30% of the total virus, an impressive result given that nef-deleted viruses are often crippled compared with wild-type viruses. Selection and fixation of escape mutants in acute HIV infection, when virus turnover is high, have been described84,85. In each case, the immunodominant epitope was altered so that it did not bind to the presenting HLA molecule.
The same escape mutation in the immunodominant Gag p24 epitope presented by HLA-B27 has been seen in more than eight different patients (ref. 86 and Goulder et al., personal communication). Study of this mutation and its selection by T cells is instructive of the whole phenomenon. First, the epitope is strongly immunodominant, and in patients with HLA-B27, T cells specific for this antigen are strongly selected. Three of four escape mutations described by Kelleher et al.87 occurred in late infection as virus levels rose, and one occurred in acute infection87. The commonest change was an arginine-to-lysine substitution that abrogates binding to the HLA-B27 molecule86; the lysine had not been seen before in B-clade Gag p24 sequences. However, it was found in B27+ patients only when there was a second change in the epitope, a leucine-to-methionine substitution87, four residues downstream. Recently, a third amino-acid change in the same conformational region of the p24 capsid has been found to be necessary (Kelleher et al., unpublished results). The mutant sites within the epitope are located on the same face of the seventh alpha-helix of the amino-terminal domain of the p24 capsid and they probably complement each other in the capsid structure. The necessity for multiple mutations could explain why these escape mutations usually occurred late in infection. This region of Gag p24 is well conserved and important in the packaging of the capsid88,90, so relatively few mutations may be compatible with viable virus90. HLA-B27 and B57, which are both associated with slow progression to AIDS, select epitopes in this region of p24. Thus, the ease of escape probably depends on the site of the epitope in the natural protein and this could account for different rates of disease progression associated with different HLA types9,10.
There is also circumstantial evidence for CTL epitope escape in cross-sectional studies where few or no sequential measurements are made (the commonest type of study). For example, Phillips et al.7 described changes occurring during ongoing infection in more than one epitope presented by HLA-B8. Goulder et al.91 found two HIV-infected, HLA-identical haemophiliac brothers who made CTL responses to totally different epitopes, arguing that this was a consequence of mutation in the normally immunodominant epitopes recognized by CTLs in one of the brothers. This resulted in a completely different pattern of response, offering one explanation for the complexity often seen in HIV-specific T-cell responses in chronic infection, where HIV-specific CTLs in patients of similar HLA type may respond to very different epitopes. These findings are consistent with evidence that escape from one CTL population is followed by a new CTL response to a new epitope92; this may weaken immune control over the virus because the subdominant CTL response is less effective, although this has not been shown clearly. More serious to the patient may be the eventual inability to make a new primary CTL responses when CD4+ T-cell help is damaged severely.
Studies in macaques have strengthened the notion that escape from CTLs is frequent and therefore important. Macaques infected with cloned SIV showed multiple mutations in the immunodominant CTL epitopes during the first few months of infection8. Up to eight mutations in Nef and Env epitopes were selected simultaneously. There was also clear selection for non-synonymous nucleotide changes and the haplotype of the major histocompatibility complex was critical in the selection process, strongly implicating CTLs8. Later, Allen et al.92 found that mutations at different epitopes could occur at different rates. They argued that the CTL response to the variable Tat epitope was strongly selective and therefore more protective than an equally strong response to a Gag p27 epitope that varied little. However, the Tat-specific response is of little value because the epitope alters so rapidly. These data in macaques establish escape mutation as a normal event in HIV infection, undermining control by CTLs.
These results also imply that HLA type can mould the virus. The immune system can respond to many epitopes, but a hierarchy of immunodominance exists that is exposed by the escape mutations93. Given that the predominant HLA types differ markedly in different populations, is it possible that the common HLA types in different HIV-exposed populations could have contributed to the generation of the different clades of HIV? The clades of virus have striking geographical distributions and infect different populations94, yet they all probably arose from the same source in Central Africa.
Other escape routes
Nef causes downregulation of HLA class I molecules at the surface of HIV-infected cells58 by re-routing the newly synthesized molecules to clathrin-coated pits for endosomal degradation as they leave the trans-Golgi network95. The effect is dependent on a sequence motif in the cytoplasmic tail of the classical HLA-A and HLA-B locus molecules; HLA-C and HLA-E do not have this motif and are not downregulated96. Thus HIV-infected cells may escape attack by HLA-A- and HLA-B-restricted CTLs. Natural killer (NK) cells express inhibitory receptors for HLA-C and HLA-E and so HIV-infected cells can also evade attack by NK cells. HIV-specific CTLs kill virus-infected cells that have failed to downregulate HLA class I and so select HLA-negative cells in vitro58. The loss of HLA from the cell surface takes about 48 hours, which may limit the effectiveness for escape, but Nef interferes with newly synthesized and HIV-peptide-loaded HLA molecules from a much earlier time. The strong CD8+ T-cell response (in terms of number of responding cells) to HIV in the blood cannot be taken as evidence that Nef-mediated HLA downregulation is unimportant, because the response in vivo could reflect cross-priming of CTLs by dendritic cells97 that take up HIV proteins from infected cells without risking HLA downregulation by Nef. However, the susceptibility of HIV-infected cells that expresses Nef to T-cell mediated lysis indicates that downregulation is not complete and the protection from CTL attack is only partial58.
Upregulation of Fas ligand is another consequence of Nef activity in infected cells98,99. Almost all HIV-specific T cells express Fas and so could be targets for killing by the FasL pathway or some other inhibitory effect.
Other means by which HIV can escape CTL attack include sequestration of infected cells in the central nervous system where T cells normally have no access. Any cells that are latently infected will also be invisible to the CTLs. Transition of virus from the R5 to the X4 type, when the co-receptor requirement is changed from CCR5 (natural receptor for the CC chemokines) to CXCR4 (natural receptor for stromal-derived factor) during disease progression, will mean that the virus becomes insensitive to inhibition by the CC chemokines released by antigen-activated T cells. Because HIV escape by epitope mutation would be more effective when target cells are escaping CTL killing than when virus entry is inhibited by chemokine competition, the virus change to the X4 type might favour CTL escape.
Overall there are more than enough mechanisms to explain why CTLs fail to control HIV (Table 2). Many of these processes, such as decreasing HLA class I expression, are used by many viruses, but others are special to HIV, particularly the effects of impaired helper T-cell function. Escape by epitope mutation has been reported for other viruses (reviewed in ref. 100) but seems to be particularly important for HIV and SIV.
Implications for vaccines
There is a desperate global need for a prophylactic AIDS vaccine, but it has been difficult to find candidate vaccines that stimulate effective neutralizing antibodies101. Induction of CD8+ T cells might offer a chance of partial or complete protection from HIV infection. CD8+ T cells cannot prevent infection of cells by HIV, but they could abort an infection before it becomes established, or contain the virus at a significantly lower level if HIV is not eliminated. A recent study of a DNA vaccine in macaques, aimed at stimulating cellular immune responses, resulted in virus levels 1,000-fold lower after SIV challenge than in unvaccinated control animals102. If the vaccine does not persist, the HIV-specific T cells may exist only as long-term memory cells, rather than as activated effectors. HIV exposure and infection would stimulate a secondary immune response that may take some days before fully active killer cells are generated. Memory CD8+ T cells can respond by releasing IFN-γ and chemokines within six hours and divide more rapidly than naive T cells103. Furthermore, their numbers are likely to be 100–1,000 times higher than HIV-specific naive T cells in a person unexposed to HIV or a vaccine37. Theoretically, therefore, a rapid response in the memory T-cell population103 could terminate the infection.
In the macaque, vaccines that stimulate CD8+ T-cell responses can partially protect against challenge with SIV104,105. Complete protection is rare, but reduction of virus load compared with controls after SIV challenge is seen consistently92,102. When the CTL response is large, the reduction in the level of virus at a set point after challenge is impressive and is accompanied by real survival advantage102. This is encouraging given that aggressive-challenge viruses are used at infecting doses that are 10–100 times those of a human sexual contact.
Further encouragement for vaccines that stimulate cellular immunity derives from the finding that some commercial sex workers in Nairobi are resistant to HIV infection. These stand out in a population in which 90% are infected with HIV. Most of these women make CD8+ T-cell responses to HIV without making serum anti-HIV antibody106. Similar induction of CTLs without antibody has been seen in macaques that have been treated with antiretroviral drugs soon after SIV challenge107,108 and these animals were protected from SIV challenge.
These findings encourage moves to start phase 1 trials in humans of vaccines that stimulate CTLs. Priming with DNA and then boosting with the same DNA construct in recombinant pox viruses stimulates strong CD8+ T-cell responses in macaques18,104,109. DNA together with IL-2 was also effective at stimulating CD8+ T-cell responses and protected against SIV challenge102. Serious attempts at vaccine development have to take a global perspective focusing on A- and C-clade vaccines — the clades that cause most infections worldwide. For developing countries a vaccine is the only real hope of controlling the devastating HIV pandemic that is sweeping sub-Saharan Africa and Asia.
Would CTL-inducing vaccines select escape mutants and therefore be ineffective? This is clearly possible110, but the vaccine-primed response might have advantages over natural infection that could minimize this problem by keeping virus levels low (Fig. 5). The number of antigen-specific precursors is far greater in vaccine recipients and they should respond with chemokine and cytokine expression much more rapidly103. The vaccines can also stimulate helper T cells and should therefore generate a more effective immune response which is better differentiated towards true CTLs. If a small amount of infecting virus can stimulate a strong secondary CTL response it is possible that the infection could be terminated, but this still has to be proven.
Conclusions
The cellular immune response to HIV is complex. The CD4+ T-cell response in HIV infection has long been known to be poor and we now know that it is damaged specifically early in primary HIV infection. Although earlier studies emphasized the strength of the CD8+ T-cell response, more recent data have questioned whether this is optimal. Not only may the CD8+ T-cell response be defective, but that impairment could facilitate selection of virus escape mutants that make it even harder for the immune system to maintain control over the virus. There is a link between the weak CD4+ T-cell response and the suboptimal CD8+ T-cell response to HIV. As the infection progresses, both T-cell responses decline further and immune control of the virus collapses. Vaccines that can stimulate both CD4+ and CD8+ T-cell responses to HIV may be able to control the virus early in infection before it causes major immune damage.
References
Walker, B. D. et al. HIV-specific cytotoxic T lymphocytes in seropositive individuals. Nature 328, 345–348 (1987).
Plata, F. et al. AIDS virus specific cytotoxic T lymphocytes in lung disorders. Nature 328, 348–351 (1987).
Ogg, G. S. et al. Quantitation of HIV-1-specific cytotoxic T lymphocytes and plasma viral RNA load. Science 279, 2103–2106 (1998).
Schmitz, J. E. et al. Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science 283, 857–860 (1999).
Jin, X. et al. Dramatic rise in plasma viremia after CD8(+) T cell depletion in simian immunodeficiency virus-infected macaques. J. Exp. Med. 189, 991–998 (1999).
Yang, O. O. et al. Suppression of human immunodeficiency virus type 1 replication by CD8+ cells: evidence for HLA class I-restricted triggering of cytolytic and noncytolytic mechanisms. J. Virol. 71, 3120–3128 (1997).
Phillips, R. E. et al. Human immunodeficiency virus genetic variation that can escape cytotoxic T cell recognition. Nature 354, 453–459 (1991).
Evans, D. T. et al. Virus-specific cytotoxic T-lymphocyte responses select for amino-acid variation in simian immunodeficiency virus Env and Nef. Nature Med. 5, 1270–1276 (1999).
Carrington, M. et al. HLA and HIV-1: heterozygote advantage and B*35-Cw*04 disadvantage. Science 283, 1748–1752 (1999).
Kaslow, R. A. et al. Influence of combinations of human major histocompatibility complex genes on the course of HIV-1 infection. Nature Med. 2, 405–411 (1996).
Riddell, S. R. & Greenberg, P. D. Principles for adoptive T cell therapy of human viral diseases. Annu. Rev. Immunol. 13, 545–586 (1995).
Koup, R. A. et al. Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. J. Virol. 68, 4650–4655 (1994).
Borrow, P., Lewicki, H., Hahn, B. H., Shaw, G. M. & Oldstone, M. B. Virus-specific CD8+ cytotoxic T-lymphocyte activity associated with control of viremia in primary human immunodeficiency virus type 1 infection. J. Virol. 68, 6103–6110 (1994).
Wilson, J. D. et al. Direct visualization of HIV-1-specific cytotoxic T lymphocytes during primary infection. AIDS 14, 225–233 (2000).
Kuroda, M. J. et al. Emergence of CTL coincides with clearance of virus during primary simian immunodeficiency virus infection in rhesus monkeys. J. Immunol. 162, 5127–5133 (1999).
Callan, M. F. C. et al. Large clonal expansions of CD8+ T cells in acute infectious mononucleosis. Nature Med. 2, 205–211 (1996).
Callan, M. F. et al. CD8(+) T-cell selection, function, and death in the primary immune response in vivo. J. Clin. Invest. 106, 1251–1261 (2000).
Hanke, T. et al. Effective induction of simian immunodeficiency virus-specific cytotoxic T lymphocytes in macaques by using a multiepitope gene and DNA prime-modified vaccinia virus Ankara boost vaccination regimen. J. Virol. 73, 7524–7532 (1999).
Moskophidis, D., Lechner, F., Pircher, H. & Zinkernagel, R. M. Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells. Nature 362, 758–761 (1993).
Pantaleo, G. et al. Major expansion of CD8+ T cells with a predominant Vb usage during the primary immune response to HIV. Nature 370, 463–467 (1994).
Pantaleo, G. et al. The qualitative nature of the primary immune response to HIV infection is a prognosticator of disease progression independent of the initial level of plasma viremia. Proc. Natl Acad. Sci. USA 94, 254–258 (1997).
Altman, J. et al. Direct visualization and phenotypic analysis of virus-specific T lymphocytes in HIV-infected individuals. Science 274, 94–96 (1996).
Kuroda, M. J. et al. Comparative analysis of cytotoxic T lymphocytes in lymph nodes and peripheral blood of simian immunodeficiency virus-infected rhesus monkeys. J. Virol. 73, 1573–1579 (1999).
Meyaard, L. et al. Programmed death of T cells in HIV-1 infection. Science 257, 217–219 (1992).
Hazenberg, M. D. et al. Increased cell division but not thymic dysfunction rapidly affects the T-cell receptor excision circle content of the naive T cell population in HIV-1 infection. Nature Med. 6, 1036–1042 (2000).
Lewis, D. E., Tang, D. S., Adu-Oppong, A., Schober, W. & Rodgers, J. R. Anergy and apoptosis in CD8+ T cells from HIV-infected persons. J. Immunol. 153, 412–420 (1994).
Gray, C. M. et al. Frequency of class I HLA-restricted anti-HIV CD8+ T cells in individuals receiving highly active antiretroviral therapy (HAART). J. Immunol. 162, 1780–1788 (1999).
Ogg, G. S. et al. Decay kinetics of human immunodeficiency virus-specific effector cytotoxic T lymphocytes after combination antiretroviral therapy. J. Virol. 73, 797–800 (1999).
Kalams, S. A. et al. Levels of human immunodeficiency virus type 1-specific cytotoxic T-lymphocyte effector and memory responses decline after suppression of viremia with highly active antiretroviral therapy. J. Virol. 73, 6721–6728 (1999).
Wilson, J. D. K. et al. Oligoclonal expansions of CD8(+) T cells in chronic HIV infection are antigen specific. J. Exp. Med. 188, 785–790 (1998).
Kalams, S. A. et al. T cell receptor usage and fine specificity of human immunodeficiency virus 1-specific cytotoxic T lymphocyte clones: analysis of quasispecies recognition reveals a dominant response directed against a minor in vivo variant. J. Exp. Med. 183, 1669–1679 (1996).
Moss, P. A. H. et al. Persistent high frequency of human immunodeficiency virus-specific cytotoxic T cells in peripheral blood of infected donors. Proc. Natl Acad. Sci. USA 92, 5773–5777 (1995).
Murali-Krishna, K. et al. Counting antigen-specific CD8 T cells: a reevaluation of bystander activation during viral infection. Immunity 8, 177–187 (1998).
Butz, E. A. & Bevan, M. J. Massive expansion of antigen-specific CD8+ T cells during an acute virus infection. Immunity 8, 167–175 (1998).
Gotch, F. M., Nixon, D. F., Alp, N., McMichael, A. J. & Borysiewicz, L. K. High frequency of memory and effector gag specific cytotoxic T lymphocytes in HIV seropositive individuals. Int. Immunol. 2, 707–712 (1990).
Tan, L. C. et al. A re-evaluation of the frequency of CD8+ T cells specific for EBV in healthy virus carriers. J. Immunol. 162, 1827–1835 (1999).
Carmichael, A., Jin, X., Sissons, P. & Borysiewicz, L. Quantitative analysis of the human immunodeficiency virus type 1 (HIV-1)-specific cytotoxic T lymphocyte (CTL) response at different stages of HIV-1 infection: differential CTL responses to HIV-1 and Epstein-Barr virus in late disease. J. Exp. Med. 177, 249–256 (1993).
Dunbar, P. R. et al. Direct isolation, phenotyping and cloning of low-frequency antigen-specific cytotoxic T lymphocytes from peripheral blood. Curr. Biol. 8, 413–416 (1998).
Pantaleo, G., Koenig, S., Baseler, M., Lane, H. C. & Fauci, A. S. Defective clonogenic potential of CD8+ T lymphocytes in patients with AIDS. Expansion in vivo of a nonclonogenic CD3+CD8+DR+CD25− T cell population. J. Immunol. 144, 1696–1704 (1990).
Goulder, P. J. et al. Functionally inert HIV-specific cytotoxic T lymphocytes do not play a major role in chronically infected adults and children. J. Exp. Med. 192, 1819–1832 (2000).
Kagi, D. et al. Cytotoxicity mediated by T cells and natural killer cells is greatly impaired in perforin-deficient mice. Nature 369, 31–37 (1994).
Guidotti, L. G. et al. Intracellular inactivation of the hepatitis B virus by cytotoxic T lymphocytes. Immunity 4, 25–36 (1996).
Guidotti, L. G. et al. Viral clearance without destruction of infected cells during acute HBV infection. Science 284, 825–829 (1999).
Ramsay, A. J., Ruby, J. & Ramshaw, I. A. A case for cytokines as effector molecules in the resolution of virus infections. Immunol. Today 14, 155–158 (1993).
Price, P. et al. Cytotoxic CD8+ T lymphocytes reactive with human immunodeficiency virus-1 produce granulocyte/macrophage colony-stimulating factor and variable amounts of interleukins 2, 3, and 4 following stimulation with the cognate epitope. Clin. Immunol. Immunopathol. 74, 100–106 (1995).
Jassoy, C. et al. Human immunodeficiency virus type 1-specific cytotoxic T lymphocytes release gamma interferon, tumor necrosis factor alpha (TNF-alpha), and TNF-beta when they encounter their target antigens. J. Virol. 67, 2844–2852 (1993).
Meylan, P. R., Guatelli, J. C., Munis, J. R., Richman, D. D. & Kornbluth, R. S. Mechanisms for the inhibition of HIV replication by interferons-α, -β, and -γ in primary human macrophages. Virology 193, 138–148 (1993).
Emilie, D., Maillot, M. C., Nicolas, J. F., Fior, R. & Galanaud, P. Antagonistic effect of interferon-gamma on tat-induced transactivation of HIV long terminal repeat. J. Biol. Chem. 267, 20565–20570 (1992).
Bollinger, R. C. et al. Cytokines from vaccine-induced HIV-1 specific cytotoxic T lymphocytes: effects on viral replication. AIDS Res. Hum. Retroviruses 9, 1067–1077 (1993).
Harrer, T., Jassoy, C., Harrer, E., Johnson, R. P. & Walker, B. D. Induction of HIV-1 replication in a chronically infected T-cell line by cytotoxic T lymphocytes. J. Acquir. Immune Defic. Syndr. 6, 865–871 (1993).
Wagner, L. et al. β-Chemokines are released from HIV-1-specific cytolytic T-cell granules complexed to proteoglycans. Nature 391, 908–911 (1998).
Price, D. A. et al. Antigen-specific release of β-chemokines by anti-HIV-1 cytotoxic T lymphocytes. Curr. Biol. 8, 355–358 (1998).
Cocchi, F. et al. Identification of RANTES, MIP-1α, and MIP-1β as the major HIV-suppressive factors produced by CD8+ T cells. Science 270, 1811–1815 (1995).
Mackewicz, C. & Levy, J. A. CD8+ cell anti-HIV activity: nonlytic suppression of virus replication. AIDS Res. Hum. Retroviruses 8, 1039–1050 (1992).
Levy, J. A., Mackewicz, C. E. & Barker, E. Controlling HIV pathogenesis: the role of noncytotoxic anti-HIV response of CD8+ T cells. Immunol. Today 17, 217–224 (1996).
Copeland, K., McKay, P. J. & Rosenthal, K. L. Suppression of activation of the HIV LTR by CD8+ cells is not lentivirus specific. AIDS Res. Hum. Retroviruses 11, 1321–1325 (1995).
Yang, O. O. et al. Efficient lysis of human immunodeficiency virus type 1-infected cells by cytotoxic T lymphocytes. J. Virol. 70, 5799–5806 (1996).
Collins, K. L., Chen, B. K., Kalams, S. A., Walker, B. D. & Baltimore, D. HIV-1 Nef protein protects infected primary cells against killing by cytotoxic T lymphocytes. Nature 391, 397–401 (1998).
Klenerman, P. et al. Cytotoxic T lymphocytes and viral turnover in HIV type 1 infection. Proc. Natl Acad. Sci. USA 93, 15323–15328 (1996).
Shankar, P., Xu, Z. & Lieberman, J. Viral-specific cytotoxic T lymphocytes lyse human immunodeficiency virus-infected primary T lymphocytes by the granule exocytosis pathway. Blood 94, 3084–3093 (1999).
Hadida, F. et al. Cutting edge: RANTES regulates Fas ligand expression and killing by HIV-specific CD8 cytotoxic T cells. J. Immunol. 163, 1105–1109 (1999).
Zajac, A. J. et al. Viral immune evasion due to persistence of activated T cells without effector function. J. Exp. Med. 188, 2205–2213 (1998).
Kalams, S. A. & Walker, B. D. The critical need for CD4 help in maintaining effective cytotoxic T lymphocyte responses. J. Exp. Med. 188, 2199–2204 (1998).
Appay, V. et al. HIV-specific CD8+ T-cells produce antiviral cytokines but are impaired in cytolytic function. J. Exp. Med. 192, 63–75 (2000).
Andersson, J. et al. Perforin is not co-expressed with granzyme A within cytotoxic granules in CD8 T lymphocytes present in lymphoid tissue during chronic HIV infection. AIDS 13, 1295–1303 (1999).
Trimble, L. A. & Lieberman, J. Circulating CD8 T lymphocytes in human immunodeficiency virus-infected individuals have impaired function and downmodulate CD3ζ, the signaling chain of the T-cell receptor complex. Blood 91, 585–594 (1998).
Hamann, D. et al. Phenotypic and functional separation of memory and effector human CD8+ T cells. J. Exp. Med. 186, 1407–1418 (1997).
Champagne, P. et al. Skewed maturation of memory HIV-1 specific CD8 T lymphocytes. Nature 410, 106–111(2001).
Clerici, M. et al. Detection of three distinct patterns of T helper cell dysfunction in asymptomatic, human immunodeficiency virus-seropositive patients. Independence of CD4+ cell numbers and clinical staging. J. Clin. Invest. 84, 1892–1899 (1989).
Rosenberg, E. S. et al. Vigorous HIV-1-specific CD4+ T-cell responses associated with control of viremia. Science 278, 1447–1450 (1997).
Oxenius, A. et al. Early highly active antiretroviral therapy for acute HIV-1 infection preserves immune function of CD8+ and CD4+ T lymphocytes. Proc. Natl Acad. Sci. USA 97, 3382–3387 (2000).
Pitcher, C. J. et al. HIV-1-specific CD4+ T cells are detectable in most individuals with active HIV-1 infection, but decline with prolonged viral suppression. Nature Med. 5, 518–525 (1999).
Geijtenbeek, T. B. et al. DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell 100, 587–597 (2000).
Pope, M., Gezelter, S., Gallo, N., Hoffman, L. & Steinman, R. M. Low levels of HIV-1 infection in cutaneous dendritic cells promote extensive viral replication upon binding to memory CD4+ T cells. J. Exp. Med. 182, 2045–2056 (1995).
Rosenberg, E. S. et al. Immune control of HIV-1 after early treatment of acute infection. Nature 407, 523–526 (2000).
Ridge, J. P., Di Rosa, F. & Matzinger, P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature 393, 474–478 (1998).
Walter, E. A. et al. Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T-cell clones from the donor. N. Engl. J. Med. 333, 1038–1044 (1995).
Brodie, S. J. et al. In vivo migration and function of transferred HIV-1-specific cytotoxic T cells. Nature Med. 5, 34–41 (1999).
Tan, R. et al. Rapid death of adoptively transferred T cells in acquired immunodeficiency syndrome. Blood 93, 1506–1510 (1999).
Kanazawa, S., Okamoto, T. & Peterlin, B. M. Tat competes with CIITA for the binding to P-TEFb and blocks the expression of MHC class II genes in HIV infection. Immunity 12, 61–70 (2000).
Wodarz et al. Proc. R. Soc. Lond. B (in the press).
McMichael, A. J. et al. Memory CD8+ T cells in HIV infection. Phil. Trans. R. Soc. Lond. B 355, 363–367 (2000).
Koenig, S. et al. Transfer of HIV-1 specific cytotoxic T lymphocytes to an AIDS patient leads to selection for mutant HIV variants and subsequent disease progression. Nature Med. 1, 330–336 (1995).
Borrow, P. et al. Antiviral pressure exerted by HIV-1-specific cytotoxic T lymphocytes (CTLs) during primary infection demonstrated by rapid selection of CTL escape virus. Nature Med. 3, 205–211 (1997).
Price, D. A. et al. Positive selection of HIV-1 cytotoxic T lymphocyte escape variants during primary infection. Proc. Natl Acad. Sci. USA 94, 1890–1895 (1997).
Goulder, P. J. et al. Late escape from an immunodominant cytotoxic T-lymphocyte response associated with progression to AIDS. Nature Med. 3, 212–217 (1997).
Kelleher, A. D. et al. Clustered mutations in HIV-1 gag are consistently required for escape from HLA-B27-restricted CTL responses. J. Exp. Med. 193, 375–386 (2001).
Berthet-Colominas, C. et al. Head-to-tail dimers and interdomain flexibility revealed by the crystal structure of HIV-1 capsid protein (p24) complexed with a monoclonal antibody Fab. EMBO J. 18, 1124–1136 (1999).
Jones, I. M. & Morikawa, Y. The molecular basis of HIV capsid assembly. Rev. Med. Virol. 8, 87–95 (1998).
Zhang, W. H., Hockley, D. J., Nermut, M. V. & Jones, I. M. Functional consequences of mutations in HIV-1 Gag p55 selected by CTL pressure. Virology 203, 101–105 (1994).
Goulder, P. J. R. et al. Patterns of immunodominance in HIV-1-specific cytotoxic T lymphocyte responses in two human histocompatibility leukocyte antigens (HLA)—identical Siblings with HLA-A*0201 are influenced by epitope mutation. J. Exp. Med. 185, 1423–1433 (1997).
Allen, T. M. et al. Tat-specific cytotoxic T lymphocytes select for SIV escape variants during resolution of primary viraemia. Nature 407, 386–390 (2000).
Nowak, M. A. et al. Antigenic oscillations and shifting immunodominance in HIV-1 infections. Nature 375, 606–611 (1995).
Janssens, W., Buve, A. & Nkengasong, J. N. The puzzle of HIV-1 subtypes in Africa. AIDS 11, 705–712 (1997).
Le Gall, S. et al. Nef interacts with the mu subunit of clathrin adaptor complexes and reveals a cryptic sorting signal in MHC I molecules. Immunity 8, 483–495 (1998).
Cohen, G. B. et al. The selective downregulation of class I major histocompatibility complex proteins by HIV-1 protects HIV-infected cells from NK cells. Immunity 10, 661–671 (1999).
Albert, M. L., Sauter, B. & Bhardwaj, N. Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs. Nature 392, 86–89 (1998).
Xu, X.-N. et al. Evasion of CTL responses by nef-dependent induction of Fas ligand expression on SIV-infected cells. J. Exp. Med. 186, 7–16 (1997).
Xu, X. N. et al. Induction of Fas ligand expression by HIV involves the interaction of Nef with the T cell receptor ζ chain. J. Exp. Med. 189, 1489–1496 (1999).
McMichael, A. J. & Bangham, C. R. M. (eds) Semin. Virol. 7, 1 (1996).
Connor, R. I. et al. Immunological and virological analyses of persons infected by human immunodeficiency virus type 1 while participating in trials of recombinant gp120 subunit vaccines. J. Virol. 72, 1552–1576 (1998).
Barouch, D. H. et al. Augmentation of immune responses to HIV-1 and simian immunodeficiency virus DNA vaccines by IL-2/Ig plasmid administration in rhesus monkeys. Proc. Natl Acad. Sci. USA 97, 4192–4197 (2000).
Tanchot, C. et al. Modifications of CD8+ T cell function during in vivo memory or tolerance induction. Immunity 8, 581–590 (1998).
Kent, S. J. et al. Enhanced T-cell immunogenicity and protective efficacy of a human immunodeficiency virus type 1 vaccine regimen consisting of consecutive priming with DNA and boosting with recombinant fowlpox virus. J. Virol. 72, 10180–10188 (1998).
Gallimore, A. et al. Early suppression of SIV replication by CD8+ nef-specific cytotoxic T cells in vaccinated macaques. Nature Med. 1, 1167–1173 (1995).
Rowland-Jones, S. L. et al. Cytotoxic T cell responses to multiple conserved HIV epitopes in HIV-resistant prostitutes in Nairobi. J. Clin. Invest. 102, 1758–1765 (1998).
Lifson, J. D. et al. Containment of simian immunodeficiency virus infection: cellular immune responses and protection from rechallenge following transient postinoculation antiretroviral treatment. J. Virol. 74, 2584–2593 (2000).
Putkonen, P., Makitalo, B., Bottiger, D., Biberfeld, G. & Thorstensson, R. Protection of human immunodeficiency virus type 2-exposed seronegative macaques from mucosal simian immunodeficiency virus transmission. J. Virol. 71, 4981–4984 (1997).
Allen, T. M. et al. Induction of AIDS virus-specific CTL activity in fresh, unstimulated peripheral blood lymphocytes from rhesus macaques vaccinated with a DNA prime/modified vaccinia virus Ankara boost regimen. J. Immunol. 164, 4968–4978 (2000).
Mortara, L. et al. Selection of virus variants and emergence of virus escape mutants after immunization with an epitope vaccine. J. Virol. 72, 1403–1410 (1998).
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
McMichael, A., Rowland-Jones, S. Cellular immune responses to HIV. Nature 410, 980–987 (2001). https://doi.org/10.1038/35073658
Issue Date:
DOI: https://doi.org/10.1038/35073658
This article is cited by
-
Dynamic Behavior of a General Stochastic HIV Model with Virus-to-Cell Infection, Cell-to-Cell Transmission, Immune Response and Distributed Delays
Journal of Nonlinear Science (2023)
-
A longitudinal analysis of immune escapes from HLA-B*13-restricted T-cell responses at early stage of CRF01_AE subtype HIV-1 infection and implications for vaccine design
BMC Immunology (2022)
-
The immune synapses reveal aberrant functions of CD8 T cells during chronic HIV infection
Nature Communications (2022)
-
The role of CD38 in HIV infection
AIDS Research and Therapy (2021)
-
Transcriptional insights into the CD8+ T cell response in mono-HIV and HCV infection
Journal of Translational Medicine (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
