
Abstract
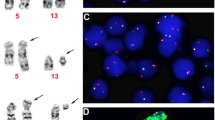
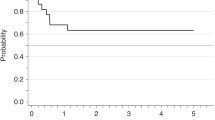
The FIP1L1-PDGFRA fusion gene has been described in patients with eosinophilia-associated myeloproliferative disorders (Eos-MPD). Here, we report on seven FIP1L1-PDGFRA-positive patients who presented with acute myeloid leukemia (AML, n=5) or lymphoblastic T-cell non-Hodgkin-lymphoma (n=2) in conjunction with AML or Eos-MPD. All patients were male, the median age was 58 years (range, 40–66). AML patients were negative for common mutations of FLT3, NRAS, NPM1, KIT, MLL and JAK2; one patient revealed a splice mutation of RUNX1 exon 7. Patients were treated with imatinib (100 mg, n=5; 400 mg, n=2) either as monotherapy (n=2), as maintenance treatment after intensive chemotherapy (n=3) or in overt relapse 43 and 72 months, respectively, after primary diagnosis and treatment of FIP1L1-PDGFRA-positive disease (n=2). All patients are alive, disease-free and in complete hematologic and complete molecular remission after a median time of 20 months (range, 9–36) on imatinib. The median time to achievement of complete molecular remission was 6 months (range, 1–14). We conclude that all eosinophilia-associated hematological malignancies should be screened for the presence of the FIP1L1-PDGFRA fusion gene as they are excellent candidates for treatment with tyrosine kinase inhibitors even if they present with an aggressive phenotype such as AML.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others

Accession codes
References
Bain BJ . Cytogenetic and molecular genetic aspects of eosinophilic leukaemias. Br J Haematol 2003; 122: 173–179.
Chusid MJ, Dale DC, West BC, Wolff SM . The hypereosinophilic syndrome: analysis of fourteen cases with review of the literature. Medicine (Baltimore) 1975; 54: 1–27.
Weller PF, Bubley GJ . The idiopathic hypereosinophilic syndrome. Blood 1994; 83: 2759–2779.
Kuroda J, Kimura S, Akaogi T, Hayashi H, Yamano T, Sasai Y et al. Myelodysplastic syndrome with clonal eosinophilia accompanied by eosinophilic pulmonary interstitial infiltration. Acta Haematol 2000; 104: 119–123.
Macdonald D, Reiter A, Cross NC . The 8p11 myeloproliferative syndrome: a distinct clinical entity caused by constitutive activation of FGFR1. Acta Haematol 2002; 107: 101–107.
Steer EJ, Cross NC . Myeloproliferative disorders with translocations of chromosome 5q31–35: role of the platelet-derived growth factor receptor beta. Acta Haematol 2002; 107: 113–122.
Cools J, DeAngelo DJ, Gotlib J, Stover EH, Legare RD, Cortes J et al. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med 2003; 348: 1201–1214.
Gilliland G, Cools J, Stover EH, Wlodarska I, Marynen P . FIP1L1-PDGFRalpha in hypereosinophilic syndrome and mastocytosis. Hematol J 2004; 5 (Suppl 3): S133–S137.
Pardanani A, Ketterling RP, Brockman SR, Flynn HC, Paternoster SF, Shearer BM et al. CHIC2 deletion, a surrogate for FIP1L1-PDGFRA fusion, occurs in systemic mastocytosis associated with eosinophilia and predicts response to imatinib mesylate therapy. Blood 2003; 102: 3093–3096.
Pardanani A, Reeder T, Porrata LF, Li CY, Tazelaar HD, Baxter EJ et al. Imatinib therapy for hypereosinophilic syndrome and other eosinophilic disorders. Blood 2003; 101: 3391–3397.
Tefferi A . Modern diagnosis and treatment of primary eosinophilia. Acta Haematol 2005; 114: 52–60.
Pardanani A, Ketterling RP, Li CY, Patnaik MM, Wolanskyj AP, Elliott MA et al. FIP1L1-PDGFRA in eosinophilic disorders: prevalence in routine clinical practice, long-term experience with imatinib therapy, and a critical review of the literature. Leuk Res 2006; 30: 965–970.
Cools J, Stover EH, Gilliland DG . Detection of the FIP1L1-PDGFRA fusion in idiopathic hypereosinophilic syndrome and chronic eosinophilic leukemia. Methods Mol Med 2006; 125: 177–187.
Robyn J, Lemery S, McCoy JP, Kubofcik J, Kim YJ, Pack S et al. Multilineage involvement of the fusion gene in patients with FIP1L1/PDGFRA-positive hypereosinophilic syndrome. Br J Haematol 2006; 132: 286–292.
Nakao M, Janssen JW, Erz D, Seriu T, Bartram CR . Tandem duplication of the FLT3 gene in acute lymphoblastic leukemia: a marker for the monitoring of minimal residual disease. Leukemia 2000; 14: 522–524.
Schnittger S, Kinkelin U, Schoch C, Heinecke A, Haase D, Haferlach T et al. Screening for MLL tandem duplication in 387 unselected patients with AML identify a prognostically unfavorable subset of AML. Leukemia 2000; 14: 796–804.
Schnittger S, Schoch C, Dugas M, Kern W, Staib P, Wuchter C et al. Analysis of FLT3 length mutations in 1003 patients with acute myeloid leukemia: correlation to cytogenetics, FAB subtype, and prognosis in the AMLCG study and usefulness as a marker for the detection of minimal residual disease. Blood 2002; 100: 59–66.
Schnittger S, Schoch C, Kern W, Mecucci C, Tschulik C, Martelli MF et al. Nucleophosmin gene mutations are predictors of favorable prognosis in acute myelogenous leukemia with a normal karyotype. Blood 2005; 106: 3733–3739.
Nakao M, Janssen JW, Seriu T, Bartram CR . Rapid and reliable detection of N-ras mutations in acute lymphoblastic leukemia by melting curve analysis using LightCycler technology. Leukemia 2000; 14: 312–315.
Miyoshi H, Ohira M, Shimizu K, Mitani K, Hirai H, Imai T et al. Alternative splicing and genomic structure of the AML1 gene involved in acute myeloid leukemia. Nucleic Acids Res 1995; 23: 2762–2769.
Score J, Curtis C, Waghorn K, Stalder M, Jotterand M, Grand FH et al. Identification of a novel imatinib responsive KIF5B-PDGFRA fusion gene following screening for PDGFRA overexpression in patients with hypereosinophilia. Leukemia 2006; 20: 827–832.
Piccaluga PP, Agostinelli C, Zinzani PL, Baccarani M, Dalla FR, Pileri SA . Expression of platelet-derived growth factor receptor alpha in peripheral T-cell lymphoma not otherwise specified. Lancet Oncol 2005; 6: 440.
Cross NC, Reiter A . Tyrosine kinase fusion genes in chronic myeloproliferative diseases. Leukemia 2002; 16: 1207–1212.
Giles FJ, Cortes JE, Kantarjian HM . Targeting the kinase activity of the BCR-ABL fusion protein in patients with chronic myeloid leukemia. Curr Mol Med 2005; 5: 615–623.
von Bubnoff N, Sandherr M, Schlimok G, Andreesen R, Peschel C, Duyster J . Myeloid blast crisis evolving during imatinib treatment of an FIP1L1-PDGFR alpha-positive chronic myeloproliferative disease with prominent eosinophilia. Leukemia 2005; 19: 286–287.
Vandenberghe P, Wlodarska I, Michaux L, Zachee P, Boogaerts M, Vanstraelen D et al. Clinical and molecular features of FIP1L1-PDFGRA (+) chronic eosinophilic leukemias. Leukemia 2004; 18: 734–742.
Mrozek K, Marcucci G, Paschka P, Whitman SP, Bloomfield CD . Clinical relevance of mutations and gene-expression changes in adult acute myeloid leukemia with normal cytogenetics: are we ready for a prognostically prioritized molecular classification? Blood 2007; 109: 431–448.
Suzuki M, Abe A, Kiyoi H, Murata M, Ito Y, Shimada K et al. Mutations of N-RAS, FLT3 and p53 genes are not involved in the development of acute leukemia transformed from myeloproliferative diseases with JAK2 mutation. Leukemia 2006; 20: 1168–1169.
Reiter A, Walz C, Watmore A, Schoch C, Blau I, Schlegelberger B et al. The t(8;9)(p22;p24) is a recurrent abnormality in chronic and acute leukemia that fuses PCM1 to JAK2. Cancer Res 2005; 65: 2662–2667.
Roche-Lestienne C, Lepers S, Soenen-Cornu V, Kahn JE, Lai JL, Hachulla E et al. Molecular characterization of the idiopathic hypereosinophilic syndrome (HES) in 35 French patients with normal conventional cytogenetics. Leukemia 2005; 19: 792–798.
Macdonald D, Aguiar RC, Mason PJ, Goldman JM, Cross NC . A new myeloproliferative disorder associated with chromosomal translocations involving 8p11: a review. Leukemia 1995; 9: 1628–1630.
Cools J, Stover EH, Boulton CL, Gotlib J, Legare RD, Amaral SM et al. PKC412 overcomes resistance to imatinib in a murine model of FIP1L1-PDGFRalpha-induced myeloproliferative disease. Cancer Cell 2003; 3: 459–469.
Lierman E, Folens C, Stover EH, Mentens N, Van Miegroet H, Scheers W et al. Sorafenib is a potent inhibitor of FIP1L1-PDGFRalpha and the imatinib-resistant FIP1L1-PDGFRalpha T674I mutant. Blood 2006; 108: 1374–1376.
Acknowledgements
This work was supported by the ‘Deutsche José Carreras Leukämie-Stiftung e.V.’ (CW, AR, Grant no. DJCLS R06/02), Germany, the Leukaemia Research Fund, United Kingdom, the Competence Network ‘Acute and chronic leukemias’, sponsored by the German Bundesministerium für Bildung und Forschung (Projektträger Gesundheitsforschung; DLR e.V. – 01GI9980/6) and the ‘European LeukemiaNet’ within the 6th European Community Framework Programme for Research and Technological Development.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Metzgeroth, G., Walz, C., Score, J. et al. Recurrent finding of the FIP1L1-PDGFRA fusion gene in eosinophilia-associated acute myeloid leukemia and lymphoblastic T-cell lymphoma. Leukemia 21, 1183–1188 (2007). https://doi.org/10.1038/sj.leu.2404662
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.leu.2404662
Keywords
This article is cited by
-
Osteolytic lesion as initial presentation in FIP1L1-PDGFRA-rearranged myeloid/lymphoid neoplasm with eosinophilia: a case report
Annals of Hematology (2024)
-
Myeloid/lymphoid neoplasms with eosinophilia and tyrosine kinase gene fusions: reevaluation of the defining characteristics in a registry-based cohort
Leukemia (2023)
-
Myeloische/lymphatische Neoplasien mit Eosinophilie und Tyrosinkinase-Fusionsgenen
Die Onkologie (2023)
-
Myeloid Neoplasm with PDGFRA Rearrangement Manifesting as a Retromolar Pad Mass
Head and Neck Pathology (2021)
