
Abstract
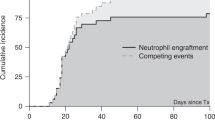
Hurler syndrome (mucopolysaccharidosis type I, MPS IH) is characterized by a deficiency of α-L-iduronidase resulting in progressive multiorgan dysfunction. We sought to determine whether enzyme replacement therapy (ERT) with iduronidase in the peritransplant period affects outcome of hematopoietic stem cell transplantation (HSCT) for MPS IH. Seven children with MPS IH at a median age of 1.5 years at the time of myeloablative HSCT were eligible. All patients had null mutations in IDUA gene. Iduronidase (0.58 mg/kg per dose) was administered intravenously in 11–14 weekly doses before HSCT and 8 weekly doses after HSCT. The infusions were well tolerated. All patients developed antibodies to iduronidase but all engrafted with >90% donor hematopoiesis. A majority of patients had significant pulmonary complications before ERT and HSCT but all are alive and well with a median follow-up of more than 1 year after HSCT. This suggests that ERT prior to HSCT is unlikely to alter engraftment. In addition, morbidity was acceptable, despite a previous history of pulmonary difficulties that suggested that these patients were high risk for these complications. Therefore, we recommend treatment of MPS IH patients with combination of ERT and HSCT therapy to further investigate its potential to enhance outcomes with HSCT.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout
Similar content being viewed by others


References
Bach G, Friedman R, Weissmann B, Neufeld EF . The defect in the Hurler and Scheie syndromes: deficiency of -L-iduronidase. Proc Natl Acad Sci USA 1972; 69: 2048–2051.
De Duve C . From cytases to lysosomes. Fed Proc 1964; 23: 1045–1049.
Boelens JJ . Trends in haematopoietic cell transplantation for inborn errors of metabolism. J Inherit Metab Dis 2006; 29: 413–420.
Fratantoni JC, Hall CW, Neufeld EF . Hurler and Hunter syndromes: mutual correction of the defect in cultured fibroblasts. Science 1968; 162: 570–572.
Fratantoni JC, Hall CW, Neufeld EF . The defect in Hurler's and Hunter's syndromes: faulty degradation of mucopolysaccharide. Proc Natl Acad Sci USA 1968; 60: 699–706.
Kakkis ED, Muenzer J, Tiller GE, Waber L, Belmont J, Passage M et al. Enzyme-replacement therapy in mucopolysaccharidosis I. N Engl J Med 2001; 344: 182–188.
Wraith JE, Clarke LA, Beck M, Kolodny EH, Pastores GM, Muenzer J et al. Enzyme replacement therapy for mucopolysaccharidosis I: a randomized, double-blinded, placebo-controlled, multinational study of recombinant human alpha-L-iduronidase (laronidase). J Pediatr 2004; 144: 581–588.
Hobbs JR, Hugh-Jones K, Barrett AJ, Byrom N, Chambers D, Henry K et al. Reversal of clinical features of Hurler's disease and biochemical improvement after treatment by bone-marrow transplantation. Lancet 1981; 2: 709–712.
Peters C, Balthazor M, Shapiro EG, King RJ, Kollman C, Hegland JD et al. Outcome of unrelated donor bone marrow transplantation in 40 children with Hurler syndrome. Blood 1996; 87: 4894–4902.
Staba SL, Escolar ML, Poe M, Kim Y, Martin PL, Szabolcs P et al. Cord-blood transplants from unrelated donors in patients with Hurler's syndrome. N Engl J Med 2004; 350: 1960–1969.
Whitley CB, Belani KG, Chang PN, Summers CG, Blazar BR, Tsai MY et al. Long-term outcome of Hurler syndrome following bone marrow transplantation. Am J Med Genet 1993; 46: 209–218.
Boelens JJ, Wynn RF, O'Meara A, Veys P, Bertrand Y, Souillet G et al. Outcomes of hematopoietic stem cell transplantation for Hurler's syndrome in Europe: a risk factor analysis for graft failure. Bone Marrow Transplant 2007; 40: 225–233.
Gassas A, Sung L, Doyle JJ, Clarke JT, Saunders EF . Life-threatening pulmonary hemorrhages post bone marrow transplantation in Hurler syndrome. Report of three cases and review of the literature. Bone Marrow Transplant 2003; 32: 213–215.
Kharbanda S, Panoskaltsis-Mortari A, Haddad IY, Blazar BR, Orchard PJ, Cornfield DN et al. Inflammatory cytokines and the development of pulmonary complications after allogeneic hematopoietic cell transplantation in patients with inherited metabolic storage disorders. Biol Blood Marrow Transplant 2006; 12: 430–437.
Cox-Brinkman J, Boelens JJ, Wraith JE, O'Meara A, Veys P, Wijburg FA et al. Haematopoietic cell transplantation (HCT) in combination with enzyme replacement therapy (ERT) in patients with Hurler syndrome. Bone Marrow Transplant 2006; 38: 17–21.
Wraith JE . Enzyme replacement therapy in mucopolysaccharidosis type I: progress and emerging difficulties. J Inherit Metab Dis 2001; 24: 245–250.
deGasperi R, Raghavan SS, Sosa MG, Kolodny EH, Carrier C, Rubenstein P et al. Measurements from normal umbilical cord blood of four lysosomal enzymatic activities: alpha-L-iduronidase (Hurler), galactocerebrosidase (globoid cell leukodystrophy), arylsulfatase A (metachromatic leukodystrophy), arylsulfatase B (Maroteaux-Lamy). Bone Marrow Transplant 2000; 25: 541–544.
Jacobson P, Park JJ, DeFor TE, Thrall M, Abel S, Krivit W et al. Oral busulfan pharmacokinetics and engraftment in children with Hurler syndrome and other inherited metabolic storage diseases undergoing hematopoietic cell transplantation. Bone Marrow Transplant 2001; 27: 855–861.
Glucksberg H, Storb R, Fefer A, Buckner CD, Neiman PE, Clift RA et al. Clinical manifestations of graft-versus-host disease in human recipients of marrow from HL-A-matched sibling donors. Transplantation 1974; 18: 295–304.
Rowlings PA, Przepiorka D, Klein JP, Gale RP, Passweg JR, Henslee-Downey PJ et al. IBMTR Severity Index for grading acute graft-versus-host disease: retrospective comparison with Glucksberg grade. Br J Haematol 1997; 97: 855–864.
Scharf SJ, Smith AG, Hansen JA, McFarland C, Erlich HA . Quantitative determination of bone marrow transplant engraftment using fluorescent polymerase chain reaction primers for human identity markers. Blood 1995; 85: 1954–1963.
Kakavanos R, Turner CT, Hopwood JJ, Kakkis ED, Brooks DA . Immune tolerance after long-term enzyme-replacement therapy among patients who have mucopolysaccharidosis I. Lancet 2003; 361: 1608–1613.
Soni S, Hente M, Breslin N, Hersh J, Whitley C, Cheerva A et al. Pre-stem cell transplantation enzyme replacement therapy in Hurler syndrome does not lead to significant antibody formation or delayed recovery of the endogenous enzyme post-transplant: a case report. Pediatr Transplant 2007; 11: 563–567.
Grewal SS, Wynn R, Abdenur JE, Burton BK, Gharib M, Haase C et al. Safety and efficacy of enzyme replacement therapy in combination with hematopoietic stem cell transplantation in Hurler syndrome. Genet Med 2005; 7: 143–146.
Sifuentes M, Doroshow R, Hoft R, Mason G, Walot I, Diament M et al. A follow-up study of MPS I patients treated with laronidase enzyme replacement therapy for 6 years. Mol Genet Metab 2007; 90: 171–180.
Peters C, Shapiro EG, Anderson J, Henslee-Downey PJ, Klemperer MR, Cowan MJ et al. Hurler syndrome: II. Outcome of HLA-genotypically identical sibling and HLA-haploidentical related donor bone marrow transplantation in fifty-four children. The storage disease collaborative study group. Blood 1998; 91: 2601–2608.
Vellodi A, Young EP, Cooper A, Wraith JE, Winchester B, Meaney C et al. Bone marrow transplantation for mucopolysaccharidosis type I: experience of two British centres. Arch Dis Child 1997; 76: 92–99.
Souillet G, Guffon N, Maire I, Pujol M, Taylor P, Sevin F et al. Outcome of 27 patients with Hurler's syndrome transplanted from either related or unrelated haematopoietic stem cell sources. Bone Marrow Transplant 2003; 31: 1105–1117.
Grewal SS, Krivit W, Defor TE, Shapiro EG, Orchard PJ, Abel SL et al. Outcome of second hematopoietic cell transplantation in Hurler syndrome. Bone Marrow Transplant 2002; 29: 491–496.
Taylor PA, Ehrhardt MJ, Roforth MM, Swedin JM, Panoskaltsis-Mortari A, Serody JS et al. Preformed antibody, not primed T cells, is the initial and major barrier to bone marrow engraftment in allosensitized recipients. Blood 2007; 109: 1307–1315.
Baxter MA, Wynn RF, Schyma L, Holmes DK, Wraith JE, Fairbairn LJ et al. Marrow stromal cells from patients affected by MPS I differentially support haematopoietic progenitor cell development. J Inherit Metab Dis 2005; 28: 1045–1053.
Acknowledgements
We thank Eileen Hanson and Teresa Kivisto for their dedication and persistence in the care of the families and in obtaining the necessary data. This study was supported by Children's Cancer Research Fund, Minneapolis, MN, USA.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tolar, J., Grewal, S., Bjoraker, K. et al. Combination of enzyme replacement and hematopoietic stem cell transplantation as therapy for Hurler syndrome. Bone Marrow Transplant 41, 531–535 (2008). https://doi.org/10.1038/sj.bmt.1705934
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bmt.1705934
Keywords
This article is cited by
-
In vivo adenine base editing corrects newborn murine model of Hurler syndrome
Molecular Biomedicine (2023)
-
Enzyme replacement therapy for mucopolysaccharidoses; past, present, and future
Journal of Human Genetics (2019)
-
Open issues in Mucopolysaccharidosis type I-Hurler
Orphanet Journal of Rare Diseases (2017)
-
Long term survival and cardiopulmonary outcome in children with Hurler syndrome after haematopoietic stem cell transplantation
Journal of Inherited Metabolic Disease (2017)
-
Increased longevity and metabolic correction following syngeneic BMT in a murine model of mucopolysaccharidosis type I
Bone Marrow Transplantation (2012)
