
Abstract
Cannabis is one of the most widely used illicit substances and there is growing interest in the association between cannabis use and psychosis. Delta-9-Tetrahydrocannabinol (Δ-9-THC) the principal active ingredient of cannabis has been shown to induce psychotomimetic and amnestic effects in healthy individuals. Whether people who frequently use cannabis are either protected from or are tolerant to these effects of Δ-9-THC has not been established. In a 3-day, double-blind, randomized, placebo-controlled study, the dose-related effects of 0, 2.5, and 5 mg intravenous Δ-9-THC were studied in 30 frequent users of cannabis and compared to 22 healthy controls. Δ-9-THC (1) produced transient psychotomimetic effects and perceptual alterations; (2) impaired memory and attention; (3) increased subjective effects of ‘high’; (4) produced tachycardia; and (5) increased serum cortisol in both groups. However, relative to controls, frequent users showed blunted responses to the psychotomimetic, perceptual altering, cognitive impairing, anxiogenic, and cortisol increasing effects of Δ-9-THC but not to its euphoric effects. Frequent users also had lower prolactin levels. These data suggest that frequent users of cannabis are either inherently blunted in their response to, and/or develop tolerance to the psychotomimetic, perceptual altering, amnestic, endocrine, and other effects of cannabinoids.
Similar content being viewed by others

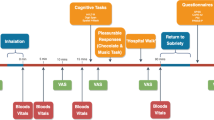
INTRODUCTION
Cannabis is one of the most widely used illicit substances and recent evidence suggests an increase in the prevalence of cannabis use, abuse, and dependence (Compton et al, 2004; SAMHSA, 2004; Stinson et al, 2006). There is considerable interest in the association between cannabis and psychosis (D'Souza, 2007; Hall et al, 2004; Henquet et al, 2005; Leweke et al, 2004; Verdoux and Tournier, 2004; Weiser and Noy, 2005). A growing number of studies suggest that the acute administration of cannabinoids including delta-9-tetrahydrocannabinol (Δ-9-THC), nabilone, and cannabis induces a broad range of transient symptoms, behaviors, and cognitive deficits in healthy individuals that resemble some aspects of endogenous psychoses (D'Souza, 2007; D'Souza et al, 2004; Henquet et al, 2006a; Leweke et al, 2004, 2000, 1999). But whether individuals who frequently use cannabis also experience such effects has not been clearly established.
While tolerance to some of the effects of cannabinoids has been reported (Green et al, 2003; Lichtman and Martin, 2005) whether tolerance develops to the psychotomimetic effects of cannabinoids is not clear. Alternatively, individuals who frequently use cannabis may be ‘protected’ from its psychotomimetic and other undesirable effects, similar to individuals at high risk for alcoholism (Schuckit, 1985a, 2000; Schuckit et al, 2004). Finally, it is unclear whether intermittent exposure to cannabis is associated with the development of tolerance (Lichtman et al, 2002).
The experimental data on cannabinoid effects is mainly based on studies of individuals with substantial exposure to cannabis. Thus, if cannabis exposure is associated with the development of tolerance or if individuals who use/abuse cannabis are protected from some of its undesirable effects, then the existing experimental literature may likely underestimate the effects of cannabinoids in cannabis naive or less experienced individuals.
METHODS
It was hypothesized that individuals who currently use cannabis frequently, hitherto referred to as frequent users, were differentially sensitive to the psychotomimetic, amnestic, perceptual altering, and endocrine effects of Δ-9-THC. This randomized, double-blind, placebo-controlled study was conducted between 1998 and 2004 at the Neurobiological Studies Unit (VA Connecticut Healthcare System (VACHS), West Haven, CT) and the Abraham Ribicoff Research Facilities (Connecticut Mental Health Center, New Haven, CT). Subjects were recruited by advertisements and by word of mouth, and were paid for their participation. The study was approved by the Protocol Review Committee of the Department of Psychiatry, Yale University School of Medicine (YUSM) and the Institutional Review Boards of both VACHS and YUSM, and was carried out in accordance with the Helsinki Declaration of 1975. Subjects were informed about the potential for adverse effects of Δ-9-THC including psychosis, anxiety, and panic.
Subjects
Current frequent users of cannabis and healthy controls were studied in parallel. Current frequent users were defined as having (1) a positive urine toxicological test for cannabis at screening, and (2) at least 10 exposures to cannabis within the past month as quantified by a time line follow back approach (Sobell and Sobell, 1992). These subjects also met criteria for current DSM-IV cannabis abuse disorder while none of the controls did. Controls were required to have (1) a negative urine toxicological test at screening, (2) no exposure to cannabis in the past week, and (3) no more than one exposure to cannabis in the past month. Data in healthy controls have been reported elsewhere (D'Souza et al, 2004).
After obtaining written informed consent, subjects (18–55 years) underwent a structured psychiatric interview for DSM-IIIR or IV (First et al, 2002) and were carefully screened for any DSM Axis I or Axis II lifetime psychiatric or substance use disorder (excluding cannabis abuse in the case of frequent users and nicotine in both groups) and family history of major Axis I disorder. All subjects were asked to estimate their lifetime cannabis exposure (number of times), heaviest exposure, and last exposure to cannabis. Cannabis-naive individuals were excluded to minimize any risk of promoting future cannabis use/abuse as were cannabis-dependent individuals. The history provided by subjects was confirmed by a telephone interview conducted with an individual (spouse or family member) identified by the subject prior to screening. A general, physical, and neurological examination, EKG and laboratory tests (serum electrolytes, liver function tests, complete blood count with differential, and urine toxicology) were also conducted. Both groups were instructed to refrain from alcohol, illicit drugs or prescription drugs not approved by the research team for 2 weeks before the study and throughout study participation. Frequent users were permitted to use cannabis until 24 h prior to each test day, to minimize cannabis withdrawal.
Subjects completed 3 test days during which they received Δ-9-THC (2.5 or 5 mg), or vehicle by intravenous (i.v.) route in a randomized, counterbalanced order under double-blind conditions (Table 1) (D'Souza et al, 2004). Staff and both groups of subjects received identical information without reference to any hypothesized group differences.
Drugs
The preparation, formulation, and storage of Δ-9-THC solution are reported elsewhere (D'Souza et al, 2004). For the control condition, an equivalent volume (≅2 ml) of ethanol (vehicle) was used which was undetectable in multiple post-injection samples. As reviewed elsewhere (D'Souza et al, 2004), the i.v route of administration, while not socially relevant, was chosen to standardize the delivery of Δ-9-THC. Subjects were administered Δ-9-THC, a point that should be noted in interpreting the results. Wachtel et al (2002), have shown that the psychoactive effects of not cannabis in healthy volunteers are primarily due to Δ-9-THC.
Test Days
Test days were separated by at least 1 week (>3 times the elimination half life of Δ-9-THC) to minimize carryover effects Table 1. Subjects fasted overnight, reported to the test facility around 0800 h, and were provided a standard breakfast. Urine toxicology was conducted on the morning of each test day to rule out recent illicit drug use. A positive urine drug screen resulted in exclusion from the study except when positive for cannabis in the frequent user group. A positive urine pregnancy test also resulted in exclusion. In-study safety procedures are described elsewhere (D'Souza et al, 2004).
Outcome Measures
Intelligence Quotient (IQ) was measured using the Slosson IQ scale (Slosson, 1963). The behavioral and cognitive outcome measures (Table 1) which were selected with a focus on psychosis, are described in detail elsewhere (D'Souza et al, 2004). Positive, negative, and general symptoms associated with schizophrenia were assessed using the Positive and Negative Syndrome Scale for Schizophrenia (PANSS) (Kay et al, 1989), perceptual alterations were measured using the Clinician Administered Dissociative Symptoms Scale (CADSS) (Bremner et al, 1998) and feeling states (‘high’, ‘calm and relaxed,’ and ‘anxiety’) associated with cannabis intoxication were measured using a self-reported visual analog scale (Haertzen, 1965, 1966). The same research coordinators rated all 3 test days for each subject. Interrater reliability sessions were conducted every 1–2 months and for example, Intraclass Correlation Coefficient for the PANSS was consistently greater than 0.85.
A cognitive test battery in a fixed sequence was initiated 30 min after Δ-9-THC administration. Unlike other measures, the cognitive battery was administered only once per test day. Verbal learning and immediate and delayed recall were measured using equivalent versions of the Hopkins Verbal Learning Test (HVLT) (Brandt, 1991; Bylsma et al, 1991). Vigilance and distractibility to visual stimuli were measured using a continuous performance task (CPT) (Gordon, 1986) in which subjects attended to numbers presented sequentially on a screen. Subjects were instructed to push a button to signal when a ‘9’ was preceded by a ‘1’. The distractibility task was identical to the vigilance task with the exception that numbers were presented sequentially in three contiguous columns. Subjects were instructed to attend to the middle column and ignore the outer two columns. Heart rate was measured continuously using a pulse oximeter. However, heart rate data was recorded for analysis as an outcome measure only at predetermined time points.
Blood was sampled from the i.v. line from the arm opposite to the one used for administering study drug (D'Souza et al, 2004) for Δ-9-THC, its primary inactive metabolite 11-nor-Δ-9-THC-9-COOH (THC-COOH), prolactin, and cortisol. Δ-9-THC and THC-COOH were only assayed from samples taken on the active Δ-9-THC test days. Endocrine measures were collected to provide biological indices of possible baseline and Δ-9-THC-induced group differences. Immediately after collection, blood samples were placed on ice, centrifuged, and the extracted plasma was alliquoted into vials for storage at −70°C until assayed. Prolactin and cortisol assays were run in duplicate pairs using antibody radioimmunoassay.
A field sobriety test was conducted at the end of each test day. The study was amended to include prospective safety assessments at 1, 3 and 6 months after the last test session to query cannabis use or the emergence of any new medical or psychiatric symptoms.
Statistical Analyses
All statistical analyses were performed in SAS Version 8.2. Baseline differences and changes from baseline were assessed in separate models. Unlike in parallel randomized controlled trials where randomization balances measured and unmeasured covariates, in this study baseline differences were expected but not of primary interest. Hence, while each measure was compared at baseline to detect baseline differences since the focus of the analysis was to detect group difference in response to Δ-9-THC, the change from baseline was of primary interest. Normal probability plots and Kolmogorov–Smirnov test statistics showed non-normality and positive skewness of the distributions of the score changes. The absence of variance during the placebo Δ-9-THC (vehicle) administration and the highly skewed responses during the Δ-9-THC conditions necessitated the use of a nonparametric approach for repeated measures data (Brunner et al, 2002). An additional advantage of this statistical approach is that it analyzes all available data on each subject including data collected on subjects who dropped out. The data were first rank-transformed and then PROC MIXED was used to fit mixed effects models with unconstrained variance–covariance structure on the ranked data. p-values for the tests of the within-subject effects were adjusted as described by Brunner et al, (2002). PANSS scores, VAS scores, CADSS clinician and CADSS subject ratings were analyzed using a nonparametric mixed model with dose (placebo, 2.5 and 5 mg) and time (P10, P80, P200) as within-subject factors and group (abuser, non-abuser) as a between-subject factor. Verbal memory (HVLT) and measures of sustained attention (CPT) were analyzed using a nonparametric mixed model with dose (placebo, 2.5, 5 mg) as a within-subject factor and group as a between-subject factor. Δ-9-THC levels were analyzed in the same way restricting the dose levels to 2.5 and 5 mg since the main interest of this analysis was to rule out pharmacokinetic differences between groups on the active Δ-9-THC conditions. Age and IQ were included as covariates in the analysis. Contrasts were used to explain significant interactions and main effects. The overall α level for each hypothesis was fixed at 0.05. Bonferroni correction was applied within but not across hypotheses. For example, for delayed recall (HVLT), a cutoff α level of 0.05/3=0.0167 was used to declare effects significant for each subscale.
For repeated measures (PANSS, CADSS, VAS), the main interaction of interest (group × dose × time) is always reported. For cognitive measures (HVLT, CPT) the main interaction of interest (group × dose) is always reported. Other interactions are reported only when the main interaction is significant. Non significant results are not reported unless otherwise specified.
RESULTS
Frequent users (n=30) and healthy control subjects (n=22) were not significantly different for age, education, socioeconomic status, or smoking status (Tables 2 and 3). However, frequent users (119±15) had significantly lower (p=0.045) IQ scores than controls (130±19), which was used as a covariate in the analysis. There were no significant group differences in dropout rates (p=0.64 Fisher's exact test).
Relative to controls, frequent users had significantly greater recent (past month) cannabis exposure and lifetime exposure to cannabis (Table 3). Further, all the frequent users reported having used cannabis sometime within 72 h prior to each test day, but not within the 24 h preceding each test day. In contrast, controls reported not having used cannabis in the week prior to each test day.
Perceptual Alterations (Cadss)
CADSS clinician-rated perceptual alterations
There were no significant baseline group differences (Figure 1). Δ-9-THC transiently increased (dose × time (ANOVA Type Statistic- ATS)=28.44, df=3.45, p<0.0001) clinician-rated perceptual alteration scores. However, frequent users had smaller Δ-9-THC-induced increases in CADSS-C scores [group by dose by time (ATS=4.79, df=3.45, p=0.001)], group by dose (ATS=2.76, df=1.18, p=0.069), and group effect (ATS=7.54, df=1, p=0.006), group by time (ATS=7.44, df=1.89, p=0.001). Post hoc analyses were conducted to compare the two groups by dose at the 10 and 80 time points. There were no significant group differences either at 10 or 80 min for placebo. However, frequent users showed significantly smaller increases relative to controls both at 10 min (ATS=4.95, df=1, p=0.0261) and 80 min (ATS=5.21, df=1, p=0.0224) for 2.5 mg Δ-9-THC. Similar differences were observed on 5 mg Δ-9-THC at both 10 min (ATS=4.05, df=1, p=0.0441) and 80 min (ATS=13.47, df=1, p=0.0002).

Perceptual alterations measured by the Clinician Administered Dissociative Symptoms Scale and psychotomimetic symptoms measured by the Positive and Negative Syndrome Scale (Tbars indicate SEMs). Frequent users had smaller Δ-9-THC induced increases in CADSS-Clinician Subscale scores and PANSS total scores relative to controls.
CADSS self-rated perceptual alterations
At baseline, frequent users reported small (mean=0.82, SD=1.58). but significantly higher scores than controls (mean=0.2, SD=0.85) (group effect: ATS=6.64, df=1, p=0.01). Δ-9-THC transiently increased self-rated perceptual alterations scores in both groups (dose × time ATS=14.64, df=3.08, p<0.0001). There was a significant group by time interaction (ATS=7.13, df=1.86, p=0.0001) but the group by dose by time interaction was not significant.
Psychotomimetic Effects
Total PANSS
There were no baseline group differences (Figure 1). Δ-9-THC transiently increased PANSS total scores in both groups (dose by time ATS=15.34, df=3.58, p<0.0001). However, frequent users had smaller increases relative to controls (group × dose × time ATS=4.34, df=3.58, p=0.0025; group by time ATS=9.34, df=1.88, p=0.0001). Post hoc comparisons for time and dose revealed that the difference between abusers and controls was significant both for the 2.5 mg dose at 10 min (ATS=6.84,df=1, p=0.0089) and 80 min (ATS=5.20, df=1, p=0.023), and the 5 mg dose at the 10 min (ATS=5.76,df=1, p=0.016), and 80 min (ATS=13.66, df=1, p=0.0002).
Self-Reported Feeling States Associated With The Cannabis Response
Visual analog scale (VAS) ‘high’
There were no significant baseline group differences (Figure 2). As expected Δ-9-THC transiently increased VAS ‘high’ scores in both groups (dose by time ATS=13.35, df=2.88, p<0.0001). While the group × dose × time interaction trended towards significance (ATS=2.48, df=2.88, p=0.06), there were no significant group (ATS=0.00, df=1, p=0.98), group by dose (ATS=0.4, df=1.93, p=0.66) or group by time (ATS=0.49, df=1.8, p=0.60) effects.

Subjective symptoms of ‘high’and anxiety measured on the Visual Analog Scale (T bars indicate SEMs). ‘high’: Δ-9-THC transiently increased scores on VAS ‘high’: equivalently in both groups. ‘anxiety’: Δ-9-THC transiently increased VAS ‘anxiety’scores but to a lower extent in frequent users compared to controls.
Visual analog scale (vas) ‘anxiety’
There were no significant baseline group differences (Figure 2). Δ-9-THC transiently increased VAS anxiety scores in both groups (dose by time ATS=5.99, df=3.32, p=0.0003). However, frequent users showed smaller increases in anxiety than controls. The group (ATS=4.04, df=1, p=0.05) and group by dose (ATS=5.44, df=1.87, p=0.005) effects were significant while the group by time (ATS=3.06, df=1.52, p=0.06) and group by dose by time (ATS=2.13, df=3.32, p=0.09) interactions showed weak trends toward significance. It is unclear why anxiety scores increased at the 200 min time point in both groups.
Visual analog scale (vas) ‘calm & relaxed’
There were no significant baseline group differences. Consistent with the above, Δ-9-THC transiently decreased VAS ‘calm and relaxed’ scores (dose × time ATS=2.42, df=3.72, p=0.05) in both groups. However, there were no group (ATS=5.47, df=1, p=0.7), group by dose (ATS=2.31, df=1.89, p=0.1) or group by dose by time (ATS=0.33, df=3.72, p=0.84) effects.
Learning And Recall(Hopkins Verbal Learning Task)
Immediate recall
Consistent with baseline differences, controls recalled more words on the placebo test day than frequent users (ATS=4.58, df=1, p=0.03), (Figure 3). As expected, Δ-9-THC impaired immediate recall in both groups in a dose-related manner (dose effect ATS=22.37, df=1.51, p<0.0001). As expected recall improved with each successive trial (trial effect ATS=166.73, df=1.86, p<0.0001). There was a significant group by dose interaction (ATS=4.06, df=1, p=0.03) with frequent users performing worse at baseline (placebo condition), yet showing smaller Δ-9-THC-induced recall impairments than controls.

Immediate and Delayed verbal recall measured by the Hopkins Verbal Learning Task (T bars indicate SEMs). Immediate recall: frequent users performed worse at baseline, but had smaller Δ-9-THC-induced impairments than controls. Delayed recall: Δ-9-THC impaired delayed recall in both groups. Only in frequent users, recall was worse on placebo than the low dose.
Delayed free recall
Δ-9-THC impaired delayed recall in both groups (dose effect (ATS=5.97, df=1.87, p=0.003)) (Figure 3). There was a significant group × dose interaction effect (ATS=4.29, df=1.87, p=0.02) with frequent users showing smaller Δ-9-THC-induced recall impairments than controls.
Delayed cued recall
While there was a group × dose interaction on cued recall (ATS=3.48, df=1.89, p=0.03), this effect did not survive Bonferroni correction.
Delayed recognition recall
Δ-9-THC did not impair recognition recall. Finally, Δ-9-THC increased the number of intrusions (dose ATS=4.48, df=1.84, p=0.013) and false-positive responses (dose ATS=9.04, df=1.96, p=0.0001) in both groups, but there were no significant group differences or group by dose interactions.
Attention
Vigilance
Δ-9-THC increased omission (dose ATS=4.11, df=1.92, p=0.02) and commission (dose ATS=3.04, df=1.98, p=0.05) errors in both groups on the vigilance task. While the group effect was not significant for both omission and commission errors, there was a significant group by dose interaction such that the difference between 5 mg and placebo dose was significant in frequent users (ATS=10.77, df=1, p=0.002 for omissions and ATS=6.91, df=1, p=0.01 for commissions) but not in controls.
Distractibility
While Δ-9-THC increased omission errors (dose ATS=6.14, df=1.57, p=0.005) in both groups, the group and group × dose interaction were not significant. There were no significant group, dose or dose × group effects on commission errors.
Heart Rate
Δ-9-THC increased heart rate in a dose dependent manner (dose: F (1,427)=65.5, p<0.0001; dose × time (F (8,427)=21.1, p<0.0001) without any significant group differences.
Plasma Δ-9-THC and 11-nor-Δ-9-THC-9-COOH (THC-COOH) levels
Plasma Δ-9-THC levels increased in a dose-dependent manner (dose: ATS=7.70, df=1.43, p=0.002) and peaked at +10 min (82 (±87) ng/dl for the 2.5 mg dose, and 119 (±166) ng/dl for the 5 mg dose). There was significant individual variability in Δ-9-THC levels. However, there were no significant group differences (ATS=0.82, df=1, p=0.36) or group by dose interactive effects on plasma Δ-9-THC levels (ATS=0.29, df=1.43, p=0.67).
As expected, relative to controls, frequent users had higher baseline plasma levels of THC-COOH the principal inactive metabolite of Δ-9-THC (ATS=105.56, df=1, p<0.0001). However, there were no significant group by dose interaction effects on plasma THC-COOH levels (ATS=1.14, df=1.53, p=0.52).
Plasma Cortisol And Prolactin
Δ-9-THC increased plasma cortisol levels in both groups (dose by time F (6,356)=5.64, p<0.0001) however, frequent users had smaller increases relative to controls (group F (1,356)=4.86, p=0.028; group × dose F (6,356)=2.5, p=0.08; group × time F (6,356)=4.6, p=0.0036; group × dose × time F (6,356)=0.6, p=0.7)) (Figure 4). Post hoc analyses revealed that controls had higher cortisol levels at the +80 (F (6,356)=7.99, p=0.005) and +140 (F (6,356)=11.75, p=0.0007) minute time points. While Δ-9-THC had no significant effects on plasma prolactin levels (dose by time: ns) in either group, frequent users had lower plasma prolactin levels (group F (1347)=15.31, p=0.0001) (Figure 4).

Plasma cortisol and prolactin levels (T bars indicate SEMs). Frequent users showed reduced Δ-9-THC-induced increases in plasma cortisol and lower overall prolactin levels.
DISCUSSION
This is the first report to our knowledge comparing the behavioral, subjective, cognitive, physiological, and endocrine effects of intravenous Δ-9-THC in frequent users of cannabis and controls.
Δ-9-THC produced a spectrum of expected behavioral, subjective, cardiovascular, and endocrine effects in both frequent users and controls. However, there were differences between the two groups. In summary, frequent users showed blunted Δ-9-THC-induced perceptual alterations (CADSS), psychotomimetic effects (PANSS), ‘anxiety’ (VAS), recall impairments, and increases in plasma cortisol. In addition, the acute effects of Δ-9-THC on several measures tended to resolve faster in frequent users as compared to controls. Frequent users also had lower baseline prolactin levels. Overall, the magnitude of the group differences in Δ-9-THC effects ranged in effect sizes of 0.38 for psychotomimetic effects (PANSS) to 0.78 for anxiety (VAS). These group differences cannot be explained by pharmacokinetic differences since there were no group differences in plasma Δ-9-THC or Δ-9-THC-COOH levels. In contrast to the above, frequent users were no different from controls in their response to Δ-9-THC-induced feeling states of ‘high’ and ‘calm and relaxed’ (VAS). Similarly, there were no group differences in the tachycardiac effects of Δ-9-THC. The finding of greater Δ-9-THC-induced commission and omission errors only on the vigilance task in frequent users is surprising since it contrasts with the general trend of frequent users showing blunted Δ-9-THC effects.
Feeling ‘high’, ‘calm and relaxed’, mellow, and creative are characterized as ‘desirable’ or positive effects of cannabis while paranoia, hallucinations, anxiety, perceptual alterations, and memory impairments are characterized as ‘undesirable’ or ‘negative’ effects (Green et al, 2003). Taken collectively, frequent users showed blunted responses to some of the ‘undesirable’ effects of Δ-9-THC but not to its ‘desirable’ effects. These group differences in Δ-9-THC effects raise the possibilities that frequent users develop tolerance to the negative effects of Δ-9-THC and/or are ‘protected’ from these effects.
Tolerance
There is considerable preclinical evidence demonstrating tolerance to most of the pharmacological effects of cannabinoids (reviewed in Gonzalez et al, 2005; Lichtman and Martin, 2005; Martin et al, 2004). However, the evidence supporting tolerance in humans is limited. Self-report (Anthony and Trinkoff, 1989), experimental (Jones et al, 1976, 1981) and direct observational (Haney et al, 1999a, 1999b) studies in humans suggest that with heavy and prolonged exposure to cannabis tolerance develops to some of its subjective and physiological effects reviewed in Lichtman and Martin, 2005. Intriguingly, Leweke et al, 2007 recently showed that frequent cannabis exposure may downregulate anandamide signaling in schizophrenic patients, but not healthy individuals. But, whether tolerance develops to the psychotomimetic and amnestic effects of cannabinoids has not been systematically studied. In light of the current focus on the association between cannabis and psychosis, this would be important. One interpretation of the current results is that frequent cannabis use is associated with the development of tolerance to the psychotomimetic effects of Δ-9-THC.
Behavioral tolerance to chronic Δ-9-THC exposure is associated with tolerance to its cerebral metabolic effects; these effects are regionally and temporally distinct (Whitlow et al, 2003). Δ-9-THC-tolerant rats also show elevated anandamide levels in limbic but not other brain regions (Di Marzo et al, 2000).
The mechanisms underlying the development of tolerance are not fully understood. What is known is that the development of tolerance is accompanied by CB1 receptor downregulation and desensitization of CB1 receptor-mediated G-protein activation. Both downregulation and desensitization develop at varying rates and magnitudes in different regions (Breivogel et al, 1999; Rodriguez de Fonseca et al, 1994; Romero et al, 1998, 1995; Rubino et al, 2000a, 2000b; Sim-Selley and Martin, 2002; Sim-Selley et al, 2006). For example, CB1 receptor downregulation and desensitization occurs faster and with greater magnitude in the hippocampus compared to the basal ganglia. In the current study, frequent users showed blunted responses to the amnestic but not to the euphoric effects of Δ-9-THC, which are believed to be mediated by different regions: the hippocampus and basal ganglia, respectively. Thus, the regional variation in the magnitude and rate of CB1 receptor adaptation may provide a possible explanation for the differential blunting of Δ-9-THC effects observed in frequent users. As reviewed by Martin et al, 2004 the precise mechanism underlying adaptation of CB1 receptor function might involve internalization of the receptor, decreased receptor synthesis, etc.
The group differences in Δ-9-THC-induced subjective, behavioral, and cognitive effects were complemented by endocrine group differences. This is the first report that we are aware of demonstrating lower prolactin levels and blunted Δ-9-THC-induced cortisol release in frequent cannabis users as compared to healthy controls. Cannabinoids increase ACTH and cortisol release via CB-1R activation in the hypothalamus pituitary (HPA) axis (Pagotto et al, 2006). The blunted Δ-9-THC-induced cortisol release in frequent users of cannabis is consistent with the animal literature (Murphy et al, 1998a). The latter is thought to reflect tolerance secondary to a downregulation of CB-1R in the HPA axis. The absence of group differences in baseline cortisol levels may be explained by the lack of very early morning (<0600 h) sampling.
Cannabinoids produce a predominantly late inhibitory effect on prolactin release (Harclerode, 1984; Murphy et al, 1998b; Pagotto et al, 2006), which is mediated by CB-1R activation of tuberoinfundibular DA neurons (Rodriguez De Fonseca et al, 1992). Δ-9-THC failed to reduce prolactin release; this may be explained by the short sampling duration. However, consistent with preclinical evidence that chronic exposure to cannabinoids leads to a long lasting suppression of prolactin release (de Miguel et al, 1998b), frequent users of cannabis had significantly lower prolactin levels compared to controls.
Frequent users had equivalent ‘high,’ ‘calm and relaxed’ feelings, and tachycardia induced by Δ-9-THC. Perhaps, as discussed earlier, tolerance to the various effects of Δ-9-THC develops at different rates reviewed in Gonzalez et al, 2005. In animals, tolerance for some effects of cannabinoids (eg, analgesia, motor inhibition hypothermia) occurs within the range of 3–7 days (Abood et al, 1993; Pertwee et al, 1993; Rubino et al, 1997), whereas the memory (Hampson et al, 2003) and endocrine effects (de Miguel et al, 1998a; Gonzalez et al, 1999), take from weeks to months to develop. These data suggest that the neural mechanisms underlying the various different brain effects of cannabinoids adapt differentially to prolonged cannabinoid exposure reviewed in Gonzalez et al, 2005. Alternatively, (frequent) users of cannabis may be innately ‘protected’ from some of the negative effects of cannabis.
Innate Differences
Several recent studies provide examples of how innate differences may account for some of the variance in the response to cannabis and also the risk for cannabis use disorders. Higher concordance in the subjective response to cannabis in monozygotic vs dizygotic twins (Lyons et al, 1997), identification of specific CB1 receptor haplotypes that contribute to the risk of developing cannabis dependence symptoms (Hopfer et al, 2006), and recent evidence of linkage for cannabis dependence on chromosome 3q21 and 9q34 (Hopfer et al, 2007) suggest genetic influences on the cannabis response. Finally, recent evidence suggests that a single nucleotide polymorphism of the catechol-methyl-transferase (COMT) gene may influence vulnerability to the psychotomimetic effects of cannabis (Caspi et al, 2005; Henquet et al, 2006b). While admittedly speculative, innate differences may contribute to the blunted ‘negative’ effects of Δ-9-THC in frequent users.
Another interpretation of the study results is that frequent users may discount negative subjective effects more than infrequent users that is, the controls in this study. While this cannot be ruled out, it is hard to extend such an explanation to the group differences in performance-based (eg, memory) and endocrine measures, which are less likely to be influenced by subjective effects.
Group Differences In Baseline And Δ-9-Thc-Induced Recall Deficits
Relative to controls, frequent users had significantly worse baseline (placebo condition) immediate, delayed, and cued recall (Figure 3). Whether these baseline differences reflect long-term or residual effects of cannabis, or innate differences is unclear. Importantly however, despite having lower IQ scores and worse recall at baseline (placebo condition), frequent users had blunted Δ-9-THC-induced immediate recall impairment relative to controls. Another intriguing finding of this study is that frequent users had better delayed recall under the influence of 2.5 mg Δ-9-THC (9.54±1.79), relative to the placebo condition (8.89±1.76) (Figure 3). While these differences were not statistically significant (ATS=1.24, df=15, p=0.27), they are consistent with and similar to other studies showing that acute exposure to cannabis normalizes the cognitive deficits associated with long-term cannabis use (Kelleher et al, 2004; Solowij, 1995, 1998). This pattern of effects is also consistent with unpublished observations in ongoing studies at our center (D'Souza et al, in review) and may represent a distinct response of frequent users to low doses of Δ-9-THC. Perhaps state (Δ-9-THC)-dependent learning or the reversal of withdrawal might explain the better performance under 2.5 mg Δ-9-THC dose in frequent users. The latter is unlikely given the absence of any baseline symptoms suggestive of withdrawal for example, nervousness, anxiety, irritability, restlessness reviewed in Budney et al, 2004 and the exclusion of cannabis dependence.
Implications For Cannabis-Related Psychosis
Individuals without any psychotic disorder, family history of psychosis or other Axis 1 disorder who frequently use cannabis may be innately protected and/or develop tolerance to the psychotomimetic and amnestic effects of Δ-9-THC. However, these data may not be relevant to individuals who have a risk for psychosis or have an established psychotic disorder.
The findings are relevant to a growing literature suggesting an association between cannabis exposure and the risk of developing a psychotic disorder. Thus, studies of individuals with significant cannabis exposure may find a lower risk for psychotic disorders, since as our data suggest these individuals either develop tolerance to or are inherently less vulnerable to the psychotomimetic effects of cannabis. Further, in association studies it may be possible that beyond a certain, albeit unspecified, magnitude of cannabis exposure, the likelihood of finding an increased risk of psychosis may actually decrease.
Implications For Cannabis Addiction
Despite the reported reinforcing effects of cannabis most people who try cannabis do not develop a cannabis use disorder (Kandel and Chen, 2000). Understanding why some but not other individuals go on to abuse cannabis is important. According to some addiction hypotheses, individuals who have either enhanced positive effects or reduced negative effects of a drug may be more likely to become addicted to it. This is perhaps best illustrated in the alcohol literature (Conrod et al, 2001; Newlin and Thomson, 1990; Pollock, 1992; Schuckit, 1985c, 1994; Schuckit et al, 1991a, 1991b, 1996). Despite similar blood alcohol levels, individuals at risk for alcoholism by family history, showed reduced consequences of alcohol administration including: (1) subjective feelings of intoxication, (2) smaller increases in body sway, (3) altered neuroendocrine responses, and (4) reduced facial flushing (Schuckit, 1985b). Follow-up studies have shown that a ‘low response’ to alcohol in this group was the strongest and most specific predictor of subsequent development of alcoholism (Schuckit, 1994). It has been hypothesized that individual who are less sensitive to some of the sedative or negative effects of alcohol, may be unable to regulate their drinking because they lack a negative feedback, a ‘brake’ on drinking, and are therefore at risk for misusing it. Similarly, individuals at risk for nicotine addiction have blunted sensitivity to the ‘negative’ effects, but heightened sensitivity to the ‘positive’ effects of nicotine (Eissenberg and Balster, 2000). Therefore, lower drug-induced negative reinforcement and either intact or higher positive reinforcement might promote the likelihood of drug abuse. Positive reactions to early cannabis use have been associated with an increased risk of later cannabis dependence (Fergusson et al, 2003b). In this study, while frequent users showed blunted responses to some of the ‘negative’ or ‘undesirable’ effects of Δ-9-THC (eg, anxiety, psychotomimetic effects), they were no different from controls in their response to some of the desirable effects of Δ-9-THC (eg, feeling ‘high’ and ‘calm & relaxed’). While admittedly speculative, we suggest that blunted responses to the negative effects of Δ-9-THC may provide an explanation as to why some individuals may be more likely to abuse cannabis (Fergusson et al, 2003a; Lyons et al, 1997).
Limitations
Perhaps a more balanced battery of assessments that included more measures of ‘positive’ effects may not have shown this profile of group differences predominantly in ‘undesirable’ effects of cannabinoids. Further, since expectancy to drug effects was not measured or manipulated it is unknown whether expectancy may have contributed to the results. However, given that participation was voluntary and that both groups had experience with cannabis, albeit to different degrees, it is unlikely that subjects had strong negative expectancy to drug effects. As discussed elsewhere (D'Souza et al, 2004), the intravenous route, the speed of drug administration, and the subjects not being able to ‘titrate’ the dose or rate of administration is different from recreational cannabis use or the substantial literature on studies with smoked and oral Δ-9-THC administration. Nevertheless, the experimental controls in the current study address some of the confounding factors associated with naturalistic studies or studies with oral/smoked Δ-9-THC (D'Souza et al, 2004). Further, this study involved the administration of Δ-9-THC and not cannabis. Cannabis consists of several compounds that may modulate Δ-9-THC effects (Hollister, 1988) and have ‘entourage’ effects (Mechoulam and Ben-Shabat, 1999; Russo and McPartland, 2003). For example, cannabidiol (CBD) may offset some Δ-9-THC effects by its anxiolytic effects (Guimaraes et al, 1994; Zuardi et al, 1982), antipsychotic-like effects (Zuardi et al, 1995, 1991), and may block the conversion of Δ-9-THC to the more psychoactive 11-hydroxy-THC (Bornheim et al, 1995). A recent clinical trial showed that stand alone cannabidiol was as effective as the gold standard antipsychotic Amisulpiride in the treatment of acutely ill schizophrenic patients (Leweke, 2007). Nevertheless, to reduce any potentially confounding effects of other cannabinoids present in herbal cannabis, only Δ-9-THC, was administered in this study. Finally, this study was not designed to discriminate the contributions of tolerance and innate differences to the group differences observed.
In summary, there are differences in the psychotomimetic, amnestic, endocrine, and subjective effects of Δ-9-THC between frequent users of cannabis and healthy controls. The precise neurobiology of these differences remains unclear and warrants further investigation. These differences may be important to consider in reviewing the existing literature on cannabinoid effects in humans. Since the latter is largely based on the study of people who use cannabis, the existing literature may underestimate the magnitude of effects of cannabinoids. These differences may also have implications for cannabis-related psychosis and addiction.
References
Abood ME, Sauss C, Fan F, Tilton CL, Martin BR (1993). Development of behavioral tolerance to delta 9-THC without alteration of cannabinoid receptor binding or mRNA levels in whole brain. Pharmacol Biochem Behav 46: 575–579.
Anthony JC, Trinkoff AM (1989). United States epidemiologic data on drug use and abuse: how are they relevant to testing abuse liability of drugs? NIDA Res Monogr 92: 241–266.
Bornheim LM, Kim KY, Li J, Perotti BY, Benet LZ (1995). Effect of cannabidiol pretreatment on the kinetics of tetrahydrocannabinol metabolites in mouse brain. Drug Metab Dispos 23: 825–831.
Brandt J (1991). ‘The hopkins verbal learning test. Development of a new memory test with 6 equivalent forms’. Clin Neuropsychol 5: 125–142.
Breivogel CS, Childers SR, Deadwyler SA, Hampson RE, Vogt LJ, Sim-Selley LJ (1999). Chronic delta9-tetrahydrocannabinol treatment produces a time-dependent loss of cannabinoid receptors and cannabinoid receptor-activated G proteins in rat brain. J Neurochem 73: 2447–2459.
Bremner JD, Krystal JH, Putnam FW, Southwick SM, Marmar C, Charney DS et al (1998). Measurement of dissociative states with the Clinician-Administered Dissociative States Scale (CADSS). J Trauma Stress 11: 125–136.
Brunner E, Domhof S, Langer F (2002). Nonparametric Analysis of Longitudinal Data in Factorial Experiments. John Wiley and Sons: New York, NY.
Budney AJ, Hughes JR, Moore BA, Vandrey R (2004). Review of the validity and significance of cannabis withdrawal syndrome. Am J Psychiatry 161: 1967–1977.
Bylsma FW, Rebok GW, Brandt J (1991). Long-term retention of implicit learning in Huntington's disease. Neuropsychologia 29: 1213–1221.
Caspi A, Moffitt TE, Cannon M, McClay J, Murray R, Harrington H et al (2005). Moderation of the effect of adolescent-onset cannabis use on adult psychosis by a functional polymorphism in the catechol-O-methyltransferase gene: longitudinal evidence of a gene X environment interaction. Biol Psychiatry 57: 1117–1127.
Compton WM, Grant BF, Colliver JD, Glantz MD, Stinson FS (2004). Prevalence of marijuana use disorders in the United States: 1991–1992 and 2001–2002. JAMA 291: 2114–2121.
Conrod PJ, Peterson JB, Pihl RO (2001). Reliability and validity of alcohol-induced heart rate increase as a measure of sensitivity to the stimulant properties of alcohol. Psychopharmacology (Berl) 157: 20–30.
de Miguel R, Romero J, Munoz RM, Garcia-Gil L, Gonzalez S, Villanua MA et al (1998a). Effects of cannabinoids on prolactin and gonadotrophin secretion: involvement of changes in hypothalamic gamma-aminobutyric acid (GABA) inputs. Biochem Pharmacol 56: 1331–1338.
de Miguel R, Romero J, Munoz RM, Garcia-Gil L, Gonzalez S, Villanua MA et al (1998b). Effects of cannabinoids on prolactin and gonadotrophin secretion: involvement of changes in hypothalamic gamma-aminobutyric acid (GABA) inputs. Biochem Pharmacol 56: 1331–1338.
Di Marzo V, Berrendero F, Bisogno T, Gonzalez S, Cavaliere P, Romero J et al (2000). Enhancement of anandamide formation in the limbic forebrain and reduction of endocannabinoid contents in the striatum of delta9-tetrahydrocannabinol-tolerant rats. J Neurochem 74: 1627–1635.
D'Souza DC, Perry E, MacDougall L, Ammerman Y, Cooper T, Wu YT et al (2004). The psychotomimetic effects of intravenous delta-9-tetrahydrocannabinol in healthy individuals: implications for psychosis. Neuropsychopharmacology 29: 1558–1572.
D'Souza DC (2007). Cannabinoids and psychosis. Int Rev Neurobiol 78: 289–326.
Eissenberg T, Balster RL (2000). Initial tobacco use episodes in children and adolescents: current knowledge, future directions. Drug Alcohol Depend 59 (Suppl 1): S41–S60.
Fergusson DM, Horwood LJ, Swain-Campbell NR (2003b). Cannabis dependence and psychotic symptoms in young people. Psychol Med 33: 15–21.
Fergusson DM, Horwood LJ, Lynskey MT, Madden PA (2003a). Early reactions to cannabis predict later dependence. Arch Gen Psychiatry 60: 1033–1039.
First MB, Spitzer RL, Gibbon M, Williams JBW (2002). Structured Clinical Interview for DSM-IV-TR Axis I Disorders Non-patient edition. In: American Psychiatric Association, Washington, DC.
Gonzalez S, Manzanares J, Berrendero F, Wenger T, Corchero J, Bisogno T et al (1999). Identification of endocannabinoids and cannabinoid CB(1) receptor mRNA in the pituitary gland. Neuroendocrinology 70: 137–145.
Gonzalez S, Cebeira M, Fernandez-Ruiz J (2005). Cannabinoid tolerance and dependence: a review of studies in laboratory animals. Pharmacol Biochem Behav 81: 300–318.
Gordon M (1986). Microprocessor-based assessment of attention deficit disorders (ADD). Psychopharmacol Bull 22: 288–290.
Green B, Kavanagh D, Young R (2003). Being stoned: a review of self-reported cannabis effects. Drug Alcohol Rev 22: 453–460.
Guimaraes FS, de Aguiar JC, Mechoulam R, Breuer A (1994). Anxiolytic effect of cannabidiol derivatives in the elevated plus-maze. Gen Pharmacol 25: 161–164.
Haertzen CA (1965). Addiction Research Center Inventory (ARCI): development of a general drug estimation scale. J Nerv Ment Dis 141: 300–307.
Haertzen CA (1966). Development of scales based on patterns of drug effects, using the addiction Research Center Inventory (ARCI). Psychol Rep 18: 163–194.
Hall W, Degenhardt L, Teesson M (2004). Cannabis use and psychotic disorders: an update. Drug Alcohol Rev 23: 433–443.
Hampson RE, Simeral JD, Kelly EJ, Deadwyler SA (2003). Tolerance to the memory disruptive effects of cannabinoids involves adaptation by hippocampal neurons. Hippocampus 13: 543–556.
Haney M, Ward AS, Comer SD, Foltin RW, Fischman MW (1999a). Abstinence symptoms following oral THC administration to humans. Psychopharmacology 141: 385–394.
Haney M, Ward AS, Comer SD, Foltin RW, Fischman MW (1999b). Abstinence symptoms following smoked marijuana in humans. Psychopharmacology (Berl) 141: 395–404.
Harclerode J (1984). Endocrine effects of marijuana in the male: preclinical studies. In: Braude MC and Ludford JP (eds). NIDA Research Monograph Series. National Institute on Drug Abuse: Rockville, MD, pp 45–114.
Henquet C, Rosa A, Krabbendam L, Papiol S, Fananas L, Drukker M et al (2006a). An experimental study of catechol-o-methyltransferase Val158Met moderation of delta-9-tetrahydrocannabinol-induced effects on psychosis and cognition. Neuropsychopharmacology 31: 2748–2757.
Henquet C, Rosa A, Krabbendam L, Papiol S, Fananás L, Drukker M et al (2006b). An experimental study of catechol-O-methyltransferase Val158Met moderation of delta-9-tetrahydrocannabinol-induced effects on psychosis and cognition. Neuropsychopharmacology 31: 2748–2757.
Henquet C, Murray R, Linszen D, van Os J (2005). The environment and schizophrenia: the role of cannabis use. Schizophr Bull 31: 608–612.
Hollister LE (1988). Cannabis--1988. Acta Psychiatr Scand Suppl 345: 108–118.
Hopfer CJ, Lessem JM, Hartman CA, Stallings MC, Cherny SS, Corley RP et al (2007). A genome-wide scan for loci influencing adolescent cannabis dependence symptoms: evidence for linkage on chromosomes 3 and 9. Drug Alcohol Depend 89: 34–41.
Hopfer CJ, Young SE, Purcell S, Crowley TJ, Stallings MC, Corley RP et al (2006). Cannabis receptor haplotype associated with fewer cannabis dependence symptoms in adolescents. Am J Med Genet B Neuropsychiatr Genet 141: 895–901.
Jones RT, Benowitz N, Bachman J (1976). Clinical studies of cannabis tolerance and dependence. Ann N Y Acad Sci 282: 221–239.
Jones RT, Benowitz NL, Herning RI (1981). Clinical relevance of cannabis tolerance and dependence. J Clin Pharmacol 21: 143S–152S.
Kandel DB, Chen K (2000). Types of marijuana users by longitudinal course. J Stud Alcohol 61: 367–378.
Kay SR, Opler LA, Lindenmayer JP (1989). The Positive and Negative Syndrome Scale (PANSS): rationale and standardisation. Br J Psychiatry-Suppl 59–67.
Kelleher LM, Stough C, Sergejew AA, Rolfe T (2004). The effects of cannabis on information-processing speed. Addict Behav 29: 1213–1219.
Leweke FM, Giuffrida A, Koethe D, Schreiber D, Nolden BM, Kranaster L et al (2007). Anandamide levels in cerebrospinal fluid of first-episode schizophrenic patients: impact of cannabis use. Schizophr Res 94: 29–36.
Leweke FM, Gerth CW, Klosterkotter J (2004). Cannabis-associated psychosis: current status of research. CNS Drugs 18: 895–910.
Leweke FM, Schneider U, Radwan M, Schmidt E, Emrich HM (2000). Different effects of nabilone and cannabidiol on binocular depth inversion in Man. Pharmacol Biochem Behav 66: 175–181.
Leweke FM, Schneider U, Thies M, Munte TF, Emrich HM (1999). Effects of synthetic delta9-tetrahydrocannabinol on binocular depth inversion of natural and artificial objects in man. Psychopharmacology (Berl) 142: 230–235.
Leweke FM (2007). Cannabidiol as an antipsychotic—new perspectives. In: 2nd International Cannabis and Mental Health Conference. Institute of Psychiatry: London, UK.
Lichtman AH, Martin BR (2005). Cannabinoid tolerance and dependence. Handb Exp Pharmacol 168: 691–717. Review.
Lichtman AH, Varvel SA, Martin BR (2002). Endocannabinoids in cognition and dependence. Prostaglandins Leukot Essent Fatty Acids 66: 269–285.
Lyons MJ, Toomey R, Meyer JM, Green AI, Eisen SA, Goldberg J et al (1997). How do genes influence marijuana use? The role of subjective effects. Addiction 92: 409–417.
Martin BR, Sim-Selley LJ, Selley DE (2004). Signaling pathways involved in the development of cannabinoid tolerance. Trends Pharmacol Sci 25: 325–330.
Mechoulam R, Ben-Shabat S (1999). From gan-zi-gun-nu to anandamide and 2-arachidonoylglycerol: the ongoing story of cannabis. Nat Prod Rep 16: 131–143.
Murphy LL, Munoz RM, Adrian BA, Villanua MA (1998a). Function of cannabinoid receptors in the neuroendocrine regulation of hormone secretion. Neurobiol Dis 5: 432–446.
Murphy LL, Munoz RM, Adrian BA, Villanua MA (1998b). Function of cannabinoid receptors in the neuroendocrine regulation of hormone secretion. Neurobiol Dis 5: 432–446.
Newlin DB, Thomson JB (1990). Alcohol challenge with sons of alcoholics: a critical review and analysis. Psychol Bull 108: 383–402.
Pagotto U, Marsicano G, Cota D, Lutz B, Pasquali R (2006). The emerging role of the endocannabinoid system in endocrine regulation and energy balance. Endocr Rev 27: 73–100.
Pertwee RG, Stevenson LA, Griffin G (1993). Cross-tolerance between delta-9-tetrahydrocannabinol and the cannabimimetic agents, CP 55,940, WIN 55,212-2 and anandamide. [erratum appears in Br J Pharmacol 1994 Mar; 111(3):968]. Br J Pharmacol 110: 1483–1490.
Pollock VE (1992). Meta-analysis of subjective sensitivity to alcohol in sons of alcoholics. Am J Psychiatry 149: 1534–1538.
Rodriguez De Fonseca F, Fernandez-Ruiz JJ, Murphy LL, Cebeira M, Steger RW, Bartke A et al (1992). Acute effects of delta-9-tetrahydrocannabinol on dopaminergic activity in several rat brain areas. Pharmacol, Biochem Behav 42: 269–275.
Rodriguez de Fonseca F, Gorriti MA, Fernandez-Ruiz JJ, Palomo T, Ramos JA (1994). Downregulation of rat brain cannabinoid binding sites after chronic delta 9-tetrahydrocannabinol treatment. Pharmacol Biochem Behav 47: 33–40.
Romero J, Berrendero F, Manzanares J, Perez A, Corchero J, Fuentes JA et al (1998). Time-course of the cannabinoid receptor down-regulation in the adult rat brain caused by repeated exposure to delta9-tetrahydrocannabinol. Synapse 30: 298–308.
Romero J, Garcia L, Fernandez-Ruiz JJ, Cebeira M, Ramos JA (1995). Changes in rat brain cannabinoid binding sites after acute or chronic exposure to their endogenous agonist, anandamide, or to delta 9-tetrahydrocannabinol. Pharmacol Biochem Behav 51: 731–737.
Rubino T, Vigano D, Massi P, Parolaro D (2000a). Changes in the cannabinoid receptor binding, G protein coupling, and cyclic AMP cascade in the CNS of rats tolerant to and dependent on the synthetic cannabinoid compound CP55,940. J Neurochem 75: 2080–2086.
Rubino T, Vigano D, Massi P, Spinello M, Zagato E, Giagnoni G et al (2000b). Chronic delta-9-tetrahydrocannabinol treatment increases cAMP levels and cAMP-dependent protein kinase activity in some rat brain regions. Neuropharmacology 39: 1331–1336.
Rubino T, Patrini G, Parenti M, Massi P, Parolaro D (1997). Chronic treatment with a synthetic cannabinoid CP-55,940 alters G-protein expression in the rat central nervous system. Brain Res Mol Brain Res 44: 191–197.
Russo EB, McPartland JM (2003). Cannabis is more than simply delta(9)-tetrahydrocannabinol. [comment]. Psychopharmacology 165: 431–432; author reply 433–434.
SAMHSA (2004). Results from the 2003 national survey on drug use and health: national findings. Substance Abuse and Mental Health Services Administration: Rockville, MD.
Schuckit MA, Greenblatt D, Gold E, Irwin M (1991b). Reactions to ethanol and diazepam in healthy young men. J Stud Alcohol 52: 180–187.
Schuckit MA, Tsuang JW, Anthenelli RM, Tipp JE, Nurnberger Jr JI (1996). Alcohol challenges in young men from alcoholic pedigrees and control families: a report from the COGA project. J Stud Alcohol 57: 368–377.
Schuckit MA, Duthie LA, Mahler HI, Irwin M, Monteiro MG (1991a). Subjective feelings and changes in body sway following diazepam in sons of alcoholics and control subjects. J Stud Alcohol 52: 601–608.
Schuckit MA, Smith TL, Anderson KG, Brown SA (2004). Testing the level of response to alcohol: social information processing model of alcoholism risk--a 20-year prospective study. Alcohol Clin Exp Res 28: 1881–1889.
Schuckit MA (1985a). Behavioral effects of alcohol in sons of alcoholics. Recent Dev Alcohol 3: 11–19.
Schuckit MA (1985c). Ethanol-induced changes in body sway in men at high alcoholism risk. Arch Gen Psychiatry 42: 375–379.
Schuckit MA (2000). Genetics of the risk for alcoholism. Am J Addict 9: 103–112.
Schuckit MA (1994). Low level of response to alcohol as a predictor of future alcoholism. Am J Psychiatry 151: 184–189.
Schuckit MA (1985b). The clinical implications of primary diagnostic groups among alcoholics. Arch Gen Psychiatry 42: 1043–1049.
Sim-Selley LJ, Martin BR (2002). Effect of chronic administration of R-(+)-[2,3-Dihydro-5-methyl-3-[(morpholinyl)methyl]pyrrolo[1,2,3-de]-1,4-benzoxazinyl]-(1-naphthalenyl)methanone mesylate (WIN55,212-2) or delta(9)-tetrahydrocannabinol on cannabinoid receptor adaptation in mice. J Pharmacol Exp Ther 303: 36–44.
Sim-Selley LJ, Schechter NS, Rorrer WK, Dalton GD, Hernandez J, Martin BR . et al (2006). Prolonged recovery rate of CB1 receptor adaptation after cessation of long-term cannabinoid administration. Mol Pharmacol 70: 986–996.
Slosson RL (1963). Slosson Intelligence Test (SIT) for Children and Adults. In: Slosson Educational Publications: East Aurora, NY.
Sobell LC, Sobell MB (1992). Timeline follow-back: a technique for assessing self-reported alcohol consumption. Litten AR, Totowa J (eds). Measuring Alcohol Consumption. The Humana Press: NJ, USA.
Solowij N (1998). Cannabis and Cognitive Functioning. Cambridge University Press: Cambridge.
Solowij N (1995). Do cognitive impairments recover following cessation of cannabis use? Life Sci 56: 2119–2126.
Stinson FS, Ruan WJ, Pickering R, Grant BF (2006). Cannabis use disorders in the USA: prevalence, correlates and co-morbidity. Psychol Med 36: 1447–1460.
Verdoux H, Tournier M (2004). Cannabis use and risk of psychosis: an etiological link? Epidemiologia e Psichiatria Sociale 13: 113–119.
Wachtel SR, ElSohly MA, Ross SA, Ambre J, de Wit H (2002). Comparison of the subjective effects of Delta(9)-tetrahydrocannabinol and marijuana in humans. Psychopharmacology (Berl) 161: 331–339.
Weiser M, Noy S (2005). Interpreting the association between cannabis use and increased risk for schizophrenia. Dialogues Clin Neurosci 7: 81–85.
Whitlow CT, Freedland CS, Porrino LJ (2003). Functional consequences of the repeated administration of Delta9-tetrahydrocannabinol in the rat. Drug Alcohol Depend 71: 169–177.
Zuardi AW, Shirakawa I, Finkelfarb E, Karniol IG (1982). Action of cannabidiol on the anxiety and other effects produced by delta 9-THC in normal subjects. Psychopharmacology 76: 245–250.
Zuardi AW, Rodrigues JA, Cunha JM (1991). Effects of cannabidiol in animal models predictive of antipsychotic activity. Psychopharmacology 104: 260–264.
Zuardi AW, Morais SL, Guimaraes FS, Mechoulam R (1995). Antipsychotic effect of cannabidiol [letter]. J Clin Psychiatry 56: 485–486.
Acknowledgements
We thank Dr George Kottayil (Unimed Pharmaceuticals Inc.) for providing Δ-9-THC. We acknowledge the critical clinical research contributions of the Biological Studies Unit, VA Connecticut Healthcare System including Elizabeth O'Donell RN; Angelina Genovese RN; Sonah Yoo RPh; Robert Sturwold RPh, and Mr Willie Ford. This study was supported by National Institute of Drug Abuse (DA12382-01) to DCD in addition to indirect support from the 1) Department of Veterans Affairs (Schizophrenia Biological Research Center, Alcohol Research Center, National Center for PTSD (John Krystal)), 2) National Institute of Mental Health (MH61019-02 to DCD).
Author information
Authors and Affiliations
Corresponding author
Additional information
CONFLICT OF INTEREST
There are no direct or indirect conflicts of interest for any of the authors relevant to the subject of this article.
Rights and permissions
About this article
Cite this article
D'Souza, D., Ranganathan, M., Braley, G. et al. Blunted Psychotomimetic and Amnestic Effects of Δ-9-Tetrahydrocannabinol in Frequent Users of Cannabis. Neuropsychopharmacol 33, 2505–2516 (2008). https://doi.org/10.1038/sj.npp.1301643
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.npp.1301643
Keywords
This article is cited by
-
Effects of acute cannabis inhalation on reaction time, decision-making, and memory using a tablet-based application
Journal of Cannabis Research (2024)
-
The Effects of Chronic Marijuana Administration on 6-OHDA-Induced Learning & Memory Impairment and Hippocampal Dopamine and Cannabinoid Receptors Interaction in Male Rats
Neurochemical Research (2023)
-
Rates and correlates of cannabis-associated psychotic symptoms in over 230,000 people who use cannabis
Translational Psychiatry (2022)
-
Sex-specific mechanisms of tolerance for the cannabinoid agonists CP55,940 and delta-9-tetrahydrocannabinol (Δ9-THC)
Psychopharmacology (2022)
-
Factors that Impact the Pharmacokinetic and Pharmacodynamic Effects of Cannabis: a Review of Human Laboratory Studies
Current Addiction Reports (2022)
