
Abstract
Ethyl-eicosapentaenoic acid (E-EPA) is an omega-3 fatty acid that has been used in a range of neuropsychiatric conditions with some benefits. However, its mechanism of action is unknown. Here, we investigate its effects on in vivo brain metabolism in first-episode psychosis (FEP). Proton magnetic resonance spectroscopy at 3 T was performed in the temporal lobes of 24 FEP patients before and after 12 weeks of treatment in the context of a larger double-blind, placebo-controlled E-EPA augmentation study. Treatment group effects for glutathione (F1,12=6.1, p=0.03), and a hemisphere-by-group interaction for glutamine/glutamate (F1,20=4.4, p=0.049) were found. Glutathione increased bilaterally and glutamate/glutamine increased in the left hemisphere following E-EPA administration. Improvement in negative symptoms correlated with metabolic brain changes, particularly glutathione (r=−0.57). These results suggest that E-EPA augmentation alters glutathione availability and modulates the glutamine/glutamate cycle in early psychosis, with some of the metabolic brain changes being correlated with negative symptom improvement. Larger confirmatory studies of these postulated metabolic brain effects of E-EPA are warranted.
Similar content being viewed by others

INTRODUCTION
Omega-3 fatty acids are essential for normal brain development, synaptic plasticity, and function (Bazan, 2005; Bazan et al, 1997). Cell biological and molecular studies suggest that omega-3 fatty acids modulate membrane fluidity (Hashimoto et al, 1999), dopaminergic (Piomelli, 1994) and serotonergic (Yao et al, 2004) neurotransmission, and differentially alter gene expression (Kitajka et al, 2002; Salvati et al, 2004). Furthermore, preclinical studies suggest that omega-3 fatty acids have neuroprotective properties (Lonergan et al, 2002; Lynch et al, 2007; Martin et al, 2002). Animal and human studies provide ample evidence that essential fatty acid deprivation during pregnancy is associated with developmental and behavioral abnormalities (Innis et al, 1999; Wainwright et al, 1994a, 1994b) that are ameliorated by essential fatty acid supplementation (Helland et al, 2003; Wainwright et al, 1994a, 1994b).
Decreased omega-3 fatty acid levels have been found in blood and postmortem brain cell membranes in several neuropsychiatric conditions (Schachter et al, 2005), in particular schizophrenia (Berger et al, 2006; Fenton et al, 2000; McNamara et al, 2007), bipolar affective disorders (Chiu et al, 2003; Hitzemann et al, 1984; Mahadik et al, 1996; Ranjekar et al, 2003), major depression (Frasure-Smith et al, 2004; McNamara et al, 2006; Mischoulon and Fava, 2000; Peet et al, 1998), and attention deficit (hyperactivity) disorder (Burgess et al, 2000; Stevens et al, 1995).
Controlled clinical trials in established schizophrenia indicate that either sole or augmentation therapy with omega-3 fatty acids may be beneficial (Emsley et al, 2002; Joy et al, 2006; Mellor et al, 1996; Peet et al, 2001), with some conflicting results (Emsley et al, 2006; Fenton et al, 2001; Peet and Horrobin, 2002a, 2002b). Furthermore, controlled clinical trials in treatment-resistant depression (Nemets et al, 2002; Peet and Horrobin, 2002a, 2002b), bipolar depression (Keck et al, 2006), bipolar affective disorder (Stoll et al, 1999), borderline personality disorder (Zanarini and Frankenburg, 2003), incarcerated young males (Gesch et al, 2002), and children with developmental coordination disorders (Richardson and Montgomery, 2005) also suggest that omega-3 fatty acids may modulate mood, impulsivity, and aggression, while potential neuroprotective effects were found in Huntington's disease (Puri et al, 2002, 2005). A recently presented, yet unpublished, study (Amminger et al, 2007) suggests that omega-3 fatty acids may delay the onset of frank psychosis in adolescents at ultra-high risk of psychotic disorders.
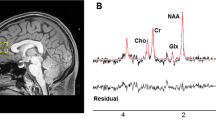
The underlying in vivo mechanisms of action of omega-3 fatty acids are still speculative. A recent study investigating T2 relaxation time the in vivo brain effects of omega-3 fatty acids using T2 relaxation time in patients with bipolar affective disorder supports preclinical findings that omega-3 fatty acids modulate membrane integrity (Hirashima et al, 2004), thereby potentially altering signal transduction and receptor binding (Vereb et al, 2003). One way to further investigate the potential in vivo brain effects of omega-3 fatty acids is proton magnetic resonance spectroscopy (1H-MRS), which can estimate regional concentrations of various brain metabolites. For example, alterations in N-acetylaspartate (NAA) most likely reflect changes in neuronal integrity, changes in glutamate/glutamine (GLX) may be linked to excitotoxicity, and changes in glutathione (GSH) related to oxidative stress (Nakamura et al, 1997) and/or apoptotic activity (Dringen, 2000). GSH synthesis is mainly regulated in astrocytes, and is important as a defense mechanism against excess NO, while functioning to protect glial and neuronal mitochondria (Gegg et al, 2003, 2005).
1H-MRS studies in established schizophrenia found altered metabolite profiles (Abbott and Bustillo, 2006; Bertolino and Weinberger, 1999; Keshavan et al, 2000; Lyoo and Renshaw, 2002; Stanley, 2002). However, 1H-MRS studies of first-episode psychosis (FEP) are less conclusive (Renshaw et al, 1995). The stage of disease at which metabolite abnormalities are established is unclear; however, studies in young, untreated FEP patients suggests that the neuronal integrity is largely preserved before onset of psychosis (Wood et al, 2003), while the majority of 1H-MRS studies in chronic schizophrenia show reduced levels of NAA.
Here, we present a study investigating the metabolic in vivo brain effects of ethyl-eicosapentaenoic acid (E-EPA) using this technique in drug-naïve or early treated FEP. We speculated that E-EPA would show neuroprotective properties in vivo by maintaining neuronal integrity, protecting the brain against excitotoxicity, and support antioxidative defense. We chose E-EPA because previous controlled studies in schizophrenia have shown some benefits (Emsley et al, 2002, 2006; Horrobin et al, 2002; Peet, 2003, 2001; Peet and Horrobin, 2002a, 2002b), and because treatment response has been associated with an increase in EPA (Arvindakshan et al, 2003).
PATIENTS AND METHODS
Patients
All study participants were patients of the Early Psychosis Prevention & Intervention Centre (EPPIC), Melbourne, Australia that covers a service area with an approximate population of 880 000. Study inclusion criteria were (1) age between 15 to 29 years (inclusive) and (2) currently psychotic as reflected by the presence of at least one psychotic symptom daily for more than 1 week (either delusions, hallucinations, disorder of thinking, and/or speech other than simple acceleration or retardation, and disorganized bizarre, or markedly inappropriate behavior). Psychotic diagnoses were confirmed using the semi-structured DSM-IV interview for research (First et al, 2002). Exclusion criteria were cases of drug-induced psychosis (self-limiting drug-related psychotic experiences that resolved within less than 7 days of drug abstinence), first episode mania, organic disorders presenting with psychotic symptoms (eg, temporal lobe epilepsy, significant neurological conditions), history of intellectual disability, or history of head injury with loss of consciousness.
Participants of this study were part of a larger randomized double-blind placebo-controlled clinical trial (Berger et al, 2008) investigating the augmenting effects of E-EPA ((5Z,8Z,11Z,14Z,17Z)-eicosa-5,8,11,14,17-pentaenoic acid) in 80 drug-naïve or early-treated FEP patients who completed repeated MRI/MRS scans. Twenty-four patients agreed to perform 1H-MRS assessments before commencement of the study medication and after 12 weeks. Twelve in each group either received 2 g oral E-EPA or 2 g placebo oil, taken as separate 1-g doses in the morning and evening (see Table 1 for details). All participants received atypical antipsychotic medication (taken nightly) according to the guidelines of the Early Psychosis Prevention & Intervention Centre, Melbourne, Australia (McGorry and Warner, 2002). Full details of the larger clinical trial design, patient characteristics, and the clinical outcome measures can be found elsewhere (Berger et al, 2008). The local research and ethics committee approved this protocol and each subject (or their guardian) provided written informed consent.
Proton Magnetic Resonance Spectroscopy
Short-echo (TE 30 ms) acquisition proton MRS was performed on a 3 T GE LX Horizon scanner (GE Healthcare, Milwaukee) using a PRESS sequence with two chemical-shift-selective imaging pulses for water suppression. Spectra were acquired with 128 transients of 2 k data points over a frequency width of 5000 Hz with TR=3 s. Spectra were recorded from single isotropic 2-cm voxels placed in each temporal lobe. Three-plane localizing images were acquired to allow prescription of regions of interest (ROI) for spectra. Sagittal plane, 2-cm thick scout images (T1 spin echo), followed by 2-cm thick coronal images, centered in the plane of the ponto-medullary junction, were acquired. An ROI in each temporal lobe was selected in the coronal plane, with the lateral aspect of the hippocampus in the center of the ROI. The sagittal image was viewed to ensure that the ROI did not include petrous temporal bone. This region of interest consisted largely of the anterior hippocampus (>50%). Spectra were analyzed with LCModel (Provencher, 1993), using a basis set of 15 metabolites acquired on-site, incorporating the standard macromolecule and baseline fitting routines of LCModel. Metabolite concentrations were estimated following calibration using the tissue water signal as an internal standard. Results are presented in institutional units approximating millimolar concentration, and were rejected if the Cramer–Rao lower bound was greater than 30%. Full-width-half-maxima and signal-to-noise ratios averaged 0.093±0.015 and 11.3±1.9, respectively, across both time points and both hemispheres. Only the following metabolites were reliably estimated in sufficient participants at both time points to allow analysis: NAA (encompassing N-acetylaspartylglutamate and N-acetylaspartate, NAA; n=24), trimethylamines (TMA; n=24), creatine/phosphocreatine (Cr/PCr; n=24), myo-inositol (mI; n=24), glutamate/glutamine (GLX; n=23), and glutathione (GSH; n=15). Metabolite concentrations were corrected for CSF and gray matter fraction within each voxel using SPM analysis of segmented T1 images (see also Supplementary Figure).
Analysis
Change scores were calculated as a percentage of the baseline metabolite concentration for each metabolite for each voxel. Repeated-measures ANCOVA (with hemisphere as the repeated measure) covarying for age was performed for each metabolite to compare differences in change between the two treatment groups. One-sample t-tests were used on the total group (collapsed across treatment) to test whether the mean change in each metabolite significantly differed from 0.
Partial correlations (controlling for age) were performed between the change scores and change in clinical variables regardless of treatment group.
RESULTS
The two treatment groups did not differ on baseline metabolite levels (all p>0.1) except for Cr/PCr (F1,19=8.2, p=0.01), where the placebo group had significantly higher concentrations than the EPA group.
No difference in percentage change between the two treatment groups was identified for TMA (F1,21=1.7, p=0.212), Cr/PCr (F1,21=2.7, p=0.115), mI (F1,21=2.5, p=0.128), or NAA (F1,21=0.0, p=0.952). However, a significant treatment group effect was found for GSH (F1,12=6.1, p=0.03) and a significant hemisphere-by-group interaction for GLX (F1,20=4.4, p=0.049). Inspection of the data (see Table 2) demonstrated that whereas the E-EPA group showed a bilateral increase in GSH, the increase in GLX was limited to the left hemisphere. When the patients with affective psychotic disorders were excluded and the analyses repeated, the effects were very similar although not quite reaching significance.
Symptom scores generally showed an improvement, ranging from a median improvement on the GAF scale of 14.5 points to a median change of 0 on the SAS. The change in the PANSS negative symptom subscale significantly correlated with three metabolites, GSH (r=−0.57, p=0.041), TMA (r=−0.48, p=0.025), and Cr/PCr (r=−0.46, p=0.032), indicating that the reduction of negative symptoms correlated strongly with percentage increase in these metabolites (see Figure 1). No other significant correlations (controlling for age) between percentage metabolite change (collapsed across hemisphere) and PANSS total, positive and general subscale scores, or CGI and GAF were identified, although the largest correlations for these latter two variables were with GSH (r=0.28 and r=−0.24 respectively). Furthermore, there were no correlations between percentage metabolite change and cumulative antipsychotic dose.

Scatterplot of the relationship between change in glutathione (GSH) and change in the PANSS negative syndrome score for both treatment groups.
Correlations between percentage change for GSH and glutamate/glutamine were positive (r=0.64, p=0.01), indicating that the changes in both metabolites were closely linked. One-sample t-tests showed no significant change in any metabolite when the two groups were combined, although the increase in NAA approached significance (t23=1.9, p=0.064; see Table 2), indicating that NAA may have increased between baseline and follow-up scan. Correlations between age and percent change were positive for all metabolites, ranging from r=0.46 (p=0.024) for NAA to r=0.33 (p=0.122) for GLX, indicating that younger age was associated with bigger metabolic changes then in older participants (see Figure 2).

Scatterplot of the relationship between change in NAA and age at scan for both treatment groups.
DISCUSSION
This study demonstrates metabolic in vivo brain effects of 2 g E-EPA on GSH and GLX in FEP, in which there was a significant increase in these metabolites in subjects receiving E-EPA augmentation treatment compared with those on treatment as usual. Further, we demonstrated that negative symptom improvement was correlated with increases in GSH, TMA, and Cr/PCr, and were not related to cumulative dose of antipsychotic medication. The moderate to strong correlation of negative symptom improvement with metabolic brain changes suggests that the E-EPA-associated increase in GSH may partially be responsible for its clinical benefits. Our findings are of particular interest in light of previous findings that glutathione levels are reduced (by 27%) in the cerebrospinal fluid of medication-free schizophrenia patients (Do et al, 2000), and postmortem brain tissue of patients with schizophrenia (Yao et al, 2006). Glutathione protects dopaminergic neurons from oxidative and excitatory damage (Grima et al, 2003; Nakamura et al, 1997). Taken together, these findings indicate that while altered GSH metabolism might play a role in schizophrenia and related disorders, augmentation therapy with E-EPA may be able to normalize this and protect dopaminergic neurons. One can speculate that the large effects of the recently presented (not yet published) omega-3 fatty acid supplementation study in prodromal adolescents (Amminger et al, 2007) may partially be explained by such a neuroprotective mechanism that may be sufficient to protect dopaminergic neurons in individuals at incipient risk for psychotic disorders.
Interestingly, the changes in GSH and GLX were positively correlated (r=0.64, p=0.01), indicating that the metabolic changes of these two metabolites were tightly coupled. This would be expected for metabolites involved in the same metabolic pathway (Dringen and Hirrlinger, 2003) when neither is at limiting concentrations for subsequent metabolic processes. Another potential explanation for the linked change in 1H-MRS signals may be that the changes occur at the site of production of both metabolites. Glutathione is mainly produced by the reaction of glutamate with cysteine and glycine, a process occurring predominantly in astrocytes (Dringen and Hirrlinger, 2003). Astrocytes are also responsible for the conversion of glutamate into glutamine (Hertz, 2004; Hertz and Zielke, 2004). Astrocytes constitute nearly half of the cells in our brain, and play a crucial role in synapse formation and functioning (Ullian et al, 2004, 2001). A relatively minor change in glial cell number (eg, via antiapoptotic mechanisms) and/or glial metabolic activity could therefore explain our findings (Berger et al, 2003; Jarskog, 2006).
The NAA increase of the combined group between baseline and follow-up scan, although only a trend, was not expected. Schizophrenia may be associated with a progressive decrease in NAA (excessive loss compared with the decrease associated with normal aging) and it has been postulated that in particular the onset of schizophrenia is associated with excessive synaptic pruning (Feinberg, 1982; Keshavan et al, 1994). However, our findings suggest that the neuronal integrity was maintained or even improved in the recovery phase of our treated FEP sample. The latter is in line with a recent study suggesting that atypical antipsychotic medication may protect the brain of first-episode schizophrenia patients from gray matter loss (Lieberman et al, 2005). The positive correlation between age and change of in vivo brain metabolites may indicate an advantage for younger FEP patients with neuroprotective treatment strategies, indicating that the potential for a restoration of synaptic integrity may decrease with adulthood (Nakamura et al, 1999; Savvateeva et al, 2000). While this would have particular importance for early intervention strategies of psychotic disorders, it needs to be confirmed by additional clinical trials.
This study has several potential limitations. First, the sample size, although similar to previous studies (Steen et al, 2005), is still relatively small, and we were not in the position to match the groups on diagnosis because the current study was embedded in a larger double-blind, placebo-controlled study of 2 g EPA augmentation in 80 drug-naïve or early treated FEP patients, limiting any stratification procedures without breaking the blindness. The results of our own larger clinical study suggest that E-EPA has more impact on non-affective psychotic disorders, accelerates treatment response, and reduces the amount of prescribed antipsychotic medication, as well as results in better tolerability (Berger et al, 2008). It is therefore noteworthy that the placebo group of the embedded Magnetic Resonance Spectroscopy study had a higher proportion of non-affective psychosis patients than the E-EPA group, meaning that we are likely to be underestimating the metabolic effect of E-EPA in the current study (as E-EPA seems to be beneficial mainly in non-affective psychosis). Second, the restriction of our ROI to the medial temporal lobes means that we cannot determine whether E-EPA has an effect throughout the brain or only in the temporal lobe. Third, the lack of a longitudinal healthy control arm means that we cannot address the question of whether the E-EPA effects were specific to early psychosis or a more general ‘healthy’ brain response (Yehuda et al, 1999). Fourth, MRS measures show normal physiological variation of between 5 and 28% (Wellard et al, 2005), meaning that it is potentially possible that our findings are merely due to normal fluctuations in metabolites. However, it seems unlikely that we would have found treatment group differences if this were the case. Finally, we had no measure of EPA adherence so it was not possible to establish a dose–response relationship. Furthermore, we did not assess dietary intake of essential fatty acids (although the dose given was 10 to 20 times the amount of EPA found in 100 g of tuna).
The MRS analysis used in this study utilizes a library of complex multipeak metabolite spectra that are matched and scaled to fit the observed subject spectrum (Provencher, 1993). Although peaks from other metabolites overlap with the glutathione spectrum, it is possible to detect contributions from metabolites with complex spectra, such as glutathione, even when the individual metabolite is not clearly visible in the spectrum (Pfeuffer et al, 1999a, 1999b). Comparisons of a short-echo single-voxel acquisition method, as used in this study, with spectral editing techniques for glutathione measurement showed that the method used in our study gives comparable measurements (Oz et al, 2006).
In conclusion, the addition of E-EPA to standard treatment in early psychosis results in a large increase in glutathione in both temporal lobes, and to a lesser extent, an increase in glutamate/glutamine that reaches significance only in the left hippocampus. Negative symptom improvements correlated with increases in the concentration of a number of metabolites, in particular glutathione. We speculate that the tightly coupled increase in GSH and GLX can be explained via a protective effect of E-EPA on astrocytes, which promotes antioxidative defense mechanisms and secures a proper functioning of the glutamate/glutamine cycle in early psychosis. Our results provide encouragement to further investigate E-EPA as a potential neuroprotective agent.
References
Abbott C, Bustillo J (2006). What have we learned from proton magnetic resonance spectroscopy about schizophrenia? A critical update. Curr Opin Psychiatry 19: 135–139.
Amminger GP, Schaefer MR, Papageorgiou K, Becker J, Mossaheb N, Harrigan S et al (2007). Omega-3 fatty acids reduce the risk of early transition to psychosis in ultra-high risk individuals: a double-blind randomized, placebo-controlled treatment study. International Conference of Schizophrenia Research (ICOSR), Colorado.
Arvindakshan M, Ghate M, Ranjekar PK, Evans DR, Mahadik SP (2003). Supplementation with a combination of omega-3 fatty acids and antioxidants (vitamins E and C) improves the outcome of schizophrenia. Schizophr Res 62: 195–204.
Bazan NG (2005). Lipid signaling in neural plasticity, brain repair, and neuroprotection. Mol Neurobiol 32: 89–103.
Bazan NG, Packard MG, Teather L, Allan G (1997). Bioactive lipids in excitatory neurotransmission and neuronal plasticity. Neurochem Int 30: 225–231.
Berger G, Wood S, McGorry P (2003). Incipient neurovulnerability and neuroprotection in early psychosis. Psychopharmacol Bull 37: 79–101.
Berger GE, Proffitt TM, McConchie MA, Yuen HP, Wood SJ, GP A et al (2008). Ethyl-eicosapentaenoic acid in first episode psychosis: a randomized, placebo-controlled trial. J Clin Psychiatry 68: 1867–1875.
Berger GE, Smesny S, Amminger GP (2006). Bioactive lipids in schizophrenia. Int Rev Psychiatry 18: 85–98.
Bertolino A, Weinberger DR (1999). Proton magnetic resonance spectroscopy in schizophrenia. Eur J Radiol 30: 132–141.
Burgess JR, Stevens L, Zhang W, Peck L (2000). Long-chain polyunsaturated fatty acids in children with attention-deficit hyperactivity disorder. Am J Clin Nutr 71: 327S–330S.
Chiu CC, Huang SY, Shen WW, Su KP (2003). Omega-3 fatty acids for depression in pregnancy. Am J Psychiatry 160: 385.
Do KQ, Trabesinger AH, Kirsten-Kruger M, Lauer CJ, Dydak U, Hell D et al (2000). Schizophrenia: glutathione deficit in cerebrospinal fluid and prefrontal cortex in vivo. Eur J Neurosci 12: 3721–3728.
Dringen R (2000). Metabolism and functions of glutathione in brain. Prog Neurobiol 62: 649–671.
Dringen R, Hirrlinger J (2003). Glutathione pathways in the brain. Biol Chem 384: 505–516.
Emsley R, Myburgh C, Oosthuizen P, van Rensburg SJ (2002). Randomized, placebo-controlled study of ethyl-eicosapentaenoic acid as supplemental treatment in schizophrenia. Am J Psychiatry 159: 1596–1598.
Emsley R, Niehaus DJ, Koen L, Oosthuizen PP, Turner HJ, Carey P et al (2006). The effects of eicosapentaenoic acid in tardive dyskinesia: a randomized, placebo-controlled trial. Schizophr Res 84: 112–120.
Feinberg I (1982). Schizophrenia: caused by a fault in programmed synaptic elimination during adolescence? J Psychiatr Res 17: 319–334.
Fenton WS, Dickerson F, Boronow J, Hibbeln JR, Knable M (2001). A placebo-controlled trial of omega-3 fatty acid (ethyl eicosapentaenoic acid) supplementation for residual symptoms and cognitive impairment in schizophrenia. Am J Psychiatry 158: 2071–2074.
Fenton WS, Hibbeln J, Knable MB (2000). Essential fatty acids, lipid membrane abnormalities, and the diagnosis and treatment of schizophrenia. Biol Psychiatry 47: 8–21.
First MB, Spitzer RL, Gibbon M, Williams JBW (2002). Structured Clinical Interview for DSM-IV-TR Axis I Disorders, Research Version, Patient Edition. (SCID-I/P). Biometrics Research, New York State Psychiatric Institute: New York.
Frasure-Smith N, Lesperance F, Julien P (2004). Major depression is associated with lower omega-3 fatty acid levels in patients with recent acute coronary syndromes. Biol Psychiatry 55: 891–896.
Gegg ME, Beltran B, Salas-Pino S, Bolanos JP, Clark JB, Moncada S et al (2003). Differential effect of nitric oxide on glutathione metabolism and mitochondrial function in astrocytes and neurones: implications for neuroprotection/neurodegeneration? J Neurochem 86: 228–237.
Gegg ME, Clark JB, Heales SJ (2005). Co-culture of neurones with glutathione deficient astrocytes leads to increased neuronal susceptibility to nitric oxide and increased glutamate–cysteine ligase activity. Brain Res 1036: 1–6.
Gesch CB, Hammond SM, Hampson SE, Eves A, Crowder MJ (2002). Influence of supplementary vitamins, minerals and essential fatty acids on the antisocial behaviour of young adult prisoners. Randomised, placebo-controlled trial. Br J Psychiatry 181: 22–28.
Grima G, Benz B, Parpura V, Cuenod M, Do KQ (2003). Dopamine-induced oxidative stress in neurons with glutathione deficit: implication for schizophrenia. Schizophr Res 62: 213–224.
Hashimoto M, Hossain S, Yamasaki H, Yazawa K, Masumura S (1999). Effects of eicosapentaenoic acid and docosahexaenoic acid on plasma membrane fluidity of aortic endothelial cells. Lipids 34: 1297–1304.
Helland IB, Smith L, Saarem K, Saugstad OD, Drevon CA (2003). Maternal supplementation with very-long-chain n-3 fatty acids during pregnancy and lactation augments children's IQ at 4 years of age. Pediatrics 111: e39–e44.
Hertz L (2004). Intercellular metabolic compartmentation in the brain: past, present and future. Neurochem Int 45: 285–296.
Hertz L, Zielke HR (2004). Astrocytic control of glutamatergic activity: astrocytes as stars of the show. Trends Neurosci 27: 735–743.
Hirashima F, Parow AM, Stoll AL, Demopulos CM, Damico KE, Rohan ML et al (2004). Omega-3 fatty acid treatment and T(2) whole brain relaxation times in bipolar disorder. Am J Psychiatry 161: 1922–1924.
Hitzemann R, Hirschowitz J, Garver D (1984). Membrane abnormalities in the psychoses and affective disorders. J Psychiatr Res 18: 319–326.
Horrobin DF, Jenkins K, Bennett CN, Christie WW (2002). Eicosapentaenoic acid and arachidonic acid: collaboration and not antagonism is the key to biological understanding. Prostaglandins Leukot Essent Fatty Acids 66: 83–90.
Innis SM, Sprecher H, Hachey D, Edmond J, Anderson RE (1999). Neonatal polyunsaturated fatty acid metabolism. Lipids 34: 139–149.
Jarskog LF (2006). Apoptosis in schizophrenia: pathophysiologic and therapeutic considerations. Curr Opin Psychiatry 19: 307–312.
Joy CB, Mumby-Croft R, Joy LA (2006). Polyunsaturated fatty acid supplementation for schizophrenia. Cochrane Database Syst Rev 3: CD001257.
Keck Jr PE, Mintz J, McElroy SL, Freeman MP, Suppes T, Frye MA et al (2006). Double-blind, randomized, placebo-controlled trials of ethyl-eicosapentanoate in the treatment of bipolar depression and rapid cycling bipolar disorder. Biol Psychiatry 60: 1020–1022.
Keshavan MS, Anderson S, Pettegrew JW (1994). Is schizophrenia due to excessive synaptic pruning in the prefrontal cortex? The Feinberg hypothesis revisited. J Psychiatr Res 28: 239–265.
Keshavan MS, Stanley JA, Pettegrew JW (2000). Magnetic resonance spectroscopy in schizophrenia: methodological issues and findings—part II. Biol Psychiatry 48: 369–380.
Kitajka K, Puskas LG, Zvara A, Hackler Jr L, Barcelo-Coblijn G, Yeo YK et al (2002). The role of n-3 polyunsaturated fatty acids in brain: modulation of rat brain gene expression by dietary n-3 fatty acids. Proc Natl Acad Sci USA 99: 2619–2624.
Lieberman JA, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, Perkins DO et al (2005). Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl Med 353: 1209–1223.
Lonergan PE, Martin DS, Horrobin DF, Lynch MA (2002). Neuroprotective effect of eicosapentaenoic acid in hippocampus of rats exposed to gamma-irradiation. J Biol Chem 277: 20804–20811.
Lynch AM, Loane DJ, Minogue AM, Clarke RM, Kilroy D, Nally RE et al (2007). Eicosapentaenoic acid confers neuroprotection in the amyloid-beta challenged aged hippocampus. Neurobiol Aging 28: 845–855.
Lyoo IK, Renshaw PF (2002). Magnetic resonance spectroscopy: current and future applications in psychiatric research. Biol Psychiatry 51: 195–207.
Mahadik SP, Mukherjee S, Horrobin DF, Jenkins K, Correnti EE, Scheffer RE (1996). Plasma membrane phospholipid fatty acid composition of cultured skin fibroblasts from schizophrenic patients: comparison with bipolar patients and normal subjects. Psychiatry Res 63: 133–142.
Martin DS, Lonergan PE, Boland B, Fogarty MP, Brady M, Horrobin DF et al (2002). Apoptotic changes in the aged brain are triggered by interleukin-1beta-induced activation of p38 and reversed by treatment with eicosapentaenoic acid. J Biol Chem 277: 34239–34246.
McGorry PD, Warner R (2002). Consensus on early intervention in schizophrenia. Schizophr Bull 28: 543–544.
McNamara RK, Hahn CG, Jandacek R, Rider T, Tso P, Stanford KE et al (2006). Selective deficits in the omega-3 fatty acid docosahexaenoic acid in the postmortem orbitofrontal cortex of patients with major depressive disorder. Biol Psychiatry 62: 17–24.
McNamara RK, Jandacek R, Rider T, Tso P, Hahn CG, Richtand NM et al (2007). Abnormalities in the fatty acid composition of the postmortem orbitofrontal cortex of schizophrenic patients: gender differences and partial normalization with antipsychotic medications. Schizophr Res 91: 37–50.
Mellor JE, Laugharne JDE, Peet M (1996). Omega-3 fatty acid supplementation in schizophrenic patients. Hum Psychopharmacol 11: 39–46.
Mischoulon D, Fava M (2000). Docosahexanoic acid and omega-3 fatty acids in depression. Psychiatr Clin North Am 23: 785–794.
Nakamura H, Kobayashi S, Ohashi Y, Ando S (1999). Age-changes of brain synapses and synaptic plasticity in response to an enriched environment. J Neurosci Res 56: 307–315.
Nakamura K, Wang W, Kang UJ (1997). The role of glutathione in dopaminergic neuronal survival. J Neurochem 69: 1850–1858.
Nemets B, Stahl Z, Belmaker RH (2002). Addition of omega-3 fatty acid to maintenance medication treatment for recurrent unipolar depressive disorder. Am J Psychiatry 159: 477–479.
Oz G, Terpstra M, Tkac I, Aia P, Lowary J, Tuite PJ et al (2006). Proton MRS of the unilateral substantia nigra in the human brain at 4 tesla: detection of high GABA concentrations. Magn Reson Med 55: 296–301.
Peet M (2003). Eicosapentaenoic acid in the treatment of schizophrenia and depression: rationale and preliminary double-blind clinical trial results. Prostaglandins Leukot Essent Fatty Acids 69: 477–485.
Peet M, Brind J, Ramchand CN, Shah SH, Vankar GK (2001). Two double-blind placebo-controlled pilot studies of eicosapentaenoic acid in the treatment of schizophrenia. Schizophr Res 49: 243–251.
Peet M, Horrobin DF (2002a). A dose-ranging exploratory study of the effects of ethyl-eicosapentaenoate in patients with persistent schizophrenic symptoms. J Psychiatr Res 36: 7–18.
Peet M, Horrobin DF (2002b). A dose-ranging study of the effects of ethyl-eicosapentaenoate in patients with ongoing depression despite apparently adequate treatment with standard drugs. Arch Gen Psych 59: 913–919.
Peet M, Murphy B, Shay J, Horrobin DF (1998). Depletion of omega-3 fatty acid levels in red blood cell membranes of depressive patients. Biol Psychiatry 43: 315–319.
Pfeuffer J, Provencher SW, Gruetter R (1999a). Water diffusion in rat brain in vivo as detected at very large b values is multicompartmental. Magma 8: 98–108.
Pfeuffer J, Tkac I, Provencher SW, Gruetter R (1999b). Toward an in vivo neurochemical profile: quantification of 18 metabolites in short-echo-time (1)H NMR spectra of the rat brain. J Magn Reson 141: 104–120.
Piomelli D (1994). Eicosanoids in synaptic transmission. Crit Rev Neurobiol 8: 65–83.
Provencher S (1993). Estimation of metabolite concentrations from localized in vivo proton NMR spectra. Magn Reson Med 30: 672–679.
Puri BK, Bydder GM, Counsell SJ, Corridan BJ, Richardson AJ, Hajnal JV et al (2002). MRI and neuropsychological improvement in Huntington disease following ethyl-EPA treatment. Neuroreport 13: 123–126.
Puri BK, Leavitt BR, Hayden MR, Ross CA, Rosenblatt A, Greenamyre JT et al (2005). Ethyl-EPA in Huntington disease: a double-blind, randomized, placebo-controlled trial. Neurology 65: 286–292.
Ranjekar PK, Hinge A, Hegde MV, Ghate M, Kale A, Sitasawad S et al (2003). Decreased antioxidant enzymes and membrane essential polyunsaturated fatty acids in schizophrenic and bipolar mood disorder patients. Psychiatry Res 121: 109–122.
Renshaw PF, Yurgelun-Todd DA, Tohen M, Gruber S, Cohen BM (1995). Temporal lobe proton magnetic resonance spectroscopy of patients with first-episode psychosis. Am J Psychiatry 152: 444–446.
Richardson AJ, Montgomery P (2005). The Oxford–Durham study: a randomized, controlled trial of dietary supplementation with fatty acids in children with developmental coordination disorder. Pediatrics 115: 1360–1366.
Salvati S, Natali F, Attorri L, Raggi C, Di Biase A, Sanchez M (2004). Stimulation of myelin proteolipid protein gene expression by eicosapentaenoic acid in C6 glioma cells. Neurochem Int 44: 331–338.
Savvateeva E, Popov A, Kamyshev N, Bragina J, Heisenberg M, Senitz D et al (2000). Age-dependent memory loss, synaptic pathology and altered brain plasticity in the Drosophila mutant cardinal accumulating 3-hydroxykynurenine. J Neural Transm 107: 581–601.
Schachter H, Kourad K, Merali Z, Lumb A, Tran K, Miguelez M et al (2005). Effects of omega-3 fatty acids on mental health. In: Quality AfHRa (ed) Evidence Report/Technology Assessment. University of Ottawa Evidence-based Practice Center: Rockville, MD, pp 1–254, under contract no. 290-02-0021.
Stanley JA (2002). In vivo magnetic resonance spectroscopy and its application to neuropsychiatric disorders. Can J Psychiatry 47: 315–326.
Steen RG, Hamer RM, Lieberman JA (2005). Measurement of brain metabolites by (1)H magnetic resonance spectroscopy in patients with schizophrenia: a systematic review and meta-analysis. Neuropsychopharmacology 30: 1949–1962.
Stevens LJ, Zentall SS, Deck JL, Abate ML, Watkins BA, Lipp SR et al (1995). Essential fatty acid metabolism in boys with attention-deficit hyperactivity disorder. Am J Clin Nutr 62: 761–768.
Stoll AL, Severus WE, Freeman MP, Rueter S, Zboyan HA, Diamond E et al (1999). Omega 3 fatty acids in bipolar disorder: a preliminary double-blind, placebo-controlled trial [see comments]. Arch Gen Psych 56: 407–412.
Ullian EM, Christopherson KS, Barres BA (2004). Role for glia in synaptogenesis. Glia 47: 209–216.
Ullian EM, Sapperstein SK, Christopherson KS, Barres BA (2001). Control of synapse number by glia. Science 291: 657–661.
Vereb G, Szollosi J, Matko J, Nagy P, Farkas T, Vigh L et al (2003). Dynamic, yet structured: the cell membrane three decades after the Singer–Nicolson model. Proc Natl Acad Sci USA 100: 8053–8058.
Wainwright PE, Huang YS, Bulman-Fleming B, Levesque S, McCutcheon D (1994a). The effects of dietary fatty acid composition combined with environmental enrichment on brain and behavior in mice. Behav Brain Res 60: 125–136.
Wainwright PE, Huang YS, Coscina DV, Levesque S, McCutcheon D (1994b). Brain and behavioral effects of dietary n-3 deficiency in mice: a three generational study. Dev Psychobiol 27: 467–487.
Wellard RM, Briellmann RS, Jennings C, Jackson GD (2005). Physiologic variability of single-voxel proton MR spectroscopic measurements at 3T. AJNR Am J Neuroradiol 26: 585–590.
Wood SJ, Berger G, Velakoulis D, Phillips LJ, McGorry PD, Yung AR et al (2003). Proton magnetic resonance spectroscopy in first episode psychosis and ultra high-risk individuals. Schizophr Bull 29: 831–843.
Yao JK, Leonard S, Reddy R (2006). Altered glutathione redox state in schizophrenia. Dis Markers 22: 83–93.
Yao JK, Magan S, Sonel AF, Gurklis JA, Sanders R, Reddy RD (2004). Effects of omega-3 fatty acid on platelet serotonin responsivity in patients with schizophrenia. Prostaglandins Leukot Essent Fatty Acids 71: 171–176.
Yehuda S, Rabinovitz S, Mostofsky DI (1999). Essential fatty acids are mediators of brain biochemistry and cognitive functions. J Neurosci Res 56: 565–570.
Zanarini MC, Frankenburg FR (2003). omega-3 fatty acid treatment of women with borderline personality disorder: a double-blind, placebo-controlled pilot study. Am J Psychiatry 160: 167–169.
Acknowledgements
A Swiss National Science Foundation young investigator award, and a Margaret & Walter Lichtenstein Foundation award (University of Basel) supported Dr Berger. A National Health & Medical Research Council Career Development Award, and a NARSAD Young Investigator Award supported Dr Stephen Wood. This work was further supported by two NH&MRC project grants (ID: 145627; 209062), a NH&MRC program grant (ID: 350241), and the Colonial Foundation, Melbourne, Australia. We want to thank all participants and their families of the Early Psychosis Prevention & Intervention Centre without whose engagement this study would not have been possible.
Author information
Authors and Affiliations
Corresponding author
Additional information
Previous presentation: Preliminary analyses have been presented at the Congress International Neuropsychopharmacology (CINP) in Paris (2004), the International Society of Magnetic Resonance in Medicine (ISMRM) meeting in Kyoto (2004), the World Federation of Societies of Biological Psychiatry (WFSBP) in Vienna (2005), and the International Congress on Schizophrenia Research in Savannah (2005).
DISCLOSURE/CONFLICT OF INTEREST
Dr Berger has no conflict of interest with any manufacturer or distributor of omega-3 fatty acids. The presented data were an investigator-initiated trial. Laxdale Ltd (now owned by Amarin Cooperations) provided the study medication for free. ORYGEN Research Centre did not receive any financial support from Laxdale Ltd. Dr Berger has received grant/research support from Astra-Zeneca, Janssen-Cilag, and Eli Lilly (not related to this trial).
Supplementary Information accompanies the paper on the Neuropsychopharmacology website (http://www.nature.com/npp)
Supplementary information
Rights and permissions
About this article
Cite this article
Berger, G., Wood, S., Wellard, R. et al. Ethyl-Eicosapentaenoic Acid in First-Episode Psychosis. A 1H-MRS Study. Neuropsychopharmacol 33, 2467–2473 (2008). https://doi.org/10.1038/sj.npp.1301628
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.npp.1301628
Keywords
This article is cited by
-
The Australian Research Council Longevity Intervention (ARCLI) study protocol (ANZCTR12611000487910) addendum: neuroimaging and gut microbiota protocol
Nutrition Journal (2019)
-
An increase in plasma brain derived neurotrophic factor levels is related to n-3 polyunsaturated fatty acid efficacy in first episode schizophrenia: secondary outcome analysis of the OFFER randomized clinical trial
Psychopharmacology (2019)
-
Focus on fatty acids in the neurometabolic pathophysiology of psychiatric disorders
Journal of Inherited Metabolic Disease (2018)
-
Erythrocyte glutathione levels as long-term predictor of transition to psychosis
Translational Psychiatry (2017)
-
Treatment for Negative Symptoms in Schizophrenia: A Comprehensive Review
Drugs (2017)

{kind=link}