
Abstract
Experiences, whether they be learning in a classroom, a stressful event, or ingestion of a psychoactive substance, impact the brain by modifying the activity and organization of specific neural circuitry. A major mechanism by which the neural activity generated by an experience modifies brain function is via modifications of synaptic transmission; that is, synaptic plasticity. Here, we review current understanding of the mechanisms of the major forms of synaptic plasticity at excitatory synapses in the mammalian brain. We also provide examples of the possible developmental and behavioral functions of synaptic plasticity and how maladaptive synaptic plasticity may contribute to neuropsychiatric disorders.
Similar content being viewed by others

INTRODUCTION
One of the most important and fascinating properties of the mammalian brain is its plasticity; the capacity of the neural activity generated by an experience to modify neural circuit function and thereby modify subsequent thoughts, feelings, and behavior. Synaptic plasticity specifically refers to the activity-dependent modification of the strength or efficacy of synaptic transmission at preexisting synapses, and for over a century has been proposed to play a central role in the capacity of the brain to incorporate transient experiences into persistent memory traces. Synaptic plasticity is also thought to play key roles in the early development of neural circuitry and evidence is accumulating that impairments in synaptic plasticity mechanisms contribute to several prominent neuropsychiatric disorders. Thus, elucidating the detailed molecular mechanisms underlying synaptic plasticity in any number of different brain regions is critical for understanding the neural basis of many aspects of normal and pathological brain function.
Given the diversity of the functions ascribed to synaptic plasticity, it is not surprising that many forms and mechanisms of synaptic plasticity have been described. Synaptic transmission can be either enhanced or depressed by activity, and these changes span temporal domains ranging from milliseconds to hours, days, and presumably even longer. Furthermore, virtually all excitatory synapses in the mammalian brain simultaneously express a number of different forms of synaptic plasticity. Here, we attempt to provide a broad overview of the mechanisms of the most prominent forms of plasticity observed at excitatory synapses in the mammalian brain. After briefly reviewing short-lasting forms of synaptic plasticity, we will emphasize current understanding of the cellular mechanisms and possible functions of the class of phenomena commonly termed long-term potentiation (LTP) and long-term depression (LTD).
SHORT-TERM SYNAPTIC PLASTICITY
Numerous forms of short-term synaptic plasticity, lasting on the order of milliseconds to several minutes, have been observed at virtually every synapse examined in organisms ranging from simple invertebrates to mammals (Zucker and Regehr, 2002). These are thought to play important roles in short-term adaptations to sensory inputs, transient changes in behavioral states, and short-lasting forms of memory. Most forms of short-term synaptic plasticity are triggered by short bursts of activity causing a transient accumulation of calcium in presynaptic nerve terminals. This increase in presynaptic calcium in turn causes changes in the probability of neurotransmitter release by directly modifying the biochemical processes that underlie the exocytosis of synaptic vesicles.
Paired-Pulse Facilitation and Depression
When two stimuli are delivered within a short interval, the response to the second stimulus can be either enhanced or depressed relative to the response to the first stimulus (Katz and Miledi, 1968; Zucker and Regehr, 2002). Paired-pulse depression is commonly observed at all synapses at short (less than 20 ms) interstimulus intervals, and most probably results from inactivation of voltage-dependent sodium or calcium channels or from a transient depletion of the release-ready pool of vesicles docked at the presynaptic terminal. Many synapses exhibit paired-pulse facilitation at longer interstimulus intervals (20–500 ms). A simple explanation for this phenomenon is that the residual calcium left over from the invasion of the first action potential contributes to additional release during the second stimulation, but it is likely that additional mechanisms are involved. These may involve activation of protein kinases that modulate the activity of presynaptic phosphoproteins. For example, mice in which the presynaptic phosphoprotein synapsin (De Camilli et al, 1990) has been knocked out, exhibit abnormal short-term plasticity (Rosahl et al, 1993, 1995).
Whether a synapse exhibits paired-pulse facilitation or depression depends on the recent history of activation of the synapse. Because these forms of plasticity largely result from changes in the probability of transmitter release (p), synapses that begin with a very high p tend to depress their response to the second pulse (Dobrunz and Stevens, 1997). In contrast, synapses with a low initial p normally exhibit an increase in p in response to the second stimulus. Consistent with this idea, manipulations that decrease p (eg, activation of presynaptic autoreceptors) almost always cause an increase in the magnitude of paired-pulse facilitation, or even a conversion of paired-pulse depression to facilitation. Thus, the same synapse can display either facilitation or depression, depending on its recent history of activation and modulation.
Facilitation and Depression Following Trains of Stimuli
Longer-lasting forms of plasticity are observed following repetitive or tetanic stimulation of synapses with prolonged (approximately 200 ms to 5 s) trains of stimulation applied at high frequencies (10–200 Hz) (Zucker and Regehr, 2002). Augmentation and post-tetanic potentiation (PTP) describe an enhancement of transmitter release lasting from seconds (augmentation) to several minutes (PTP). They also involve an increase in the probability of transmitter release in response to an action potential due, in large part, to the buildup of calcium concentration in the presynaptic terminal during the stimulus trains. This residual calcium may combine with the calcium influx elicited by the subsequent single action potential to enhance directly the release of neurotransmitter, or may lead to biochemical modifications of proteins in the presynaptic terminal (Magleby and Zengel, 1982; Zucker and Regehr, 2002).
At some synapses, repetitive activation leads to depression that can last for several seconds or even minutes (Betz, 1970; Zucker and Regehr, 2002). As in paired-pulse depression, this generally occurs in synapses that exhibit a high probability of release, and is thought to result, at least in part, from a transient depletion of the release-ready pool of synaptic vesicles. The decrease in synaptic strength can also arise from the release of modulatory substances from the activated presynaptic terminals, postsynaptic cells, or even neighboring cells, initiating a signaling cascade that leads to inhibition of the presynaptic release machinery. Finally, a postsynaptic mechanism of short-term plasticity may involve desensitization of ligand-gated receptors, making the target neuron less sensitive to neurotransmitter. A key characteristic of depression at many synapses is use dependence. Higher levels of transmission are associated with larger depression, and reduction of baseline transmission (eg, by reducing external calcium concentration) relieves depression.
Modulation of Transmission by Presynaptic Receptors
Most presynaptic terminals possess a number of different types of metabotropic G-protein-coupled receptors, as well as ionotropic receptors (MacDermott et al, 1999). The probability of transmitter release, a significant factor in defining synaptic strength, is controlled in part by the occupancy of these receptors, which in turn is set by the extracellular concentrations of their agonists. In some cases, tonic levels of endogenous ligands are sufficient to partially activate the receptors. Nonetheless, synaptic activity can further increase receptor occupancy by transiently elevating the concentration of various presynaptic neuromodulators (Thompson et al, 1993; Miller, 1998). Depending on their specific properties, activation of these receptors can either enhance or depress synaptic transmission.
Via the release of a number of different neuromodulators, postsynaptic cells can also influence the release of transmitter from presynaptic terminals. A common scenario is that in response to strong postsynaptic depolarization, dendrites release retrograde messengers that act through G-protein-coupled receptors located on presynaptic terminals to influence neurotransmitter release. Retrograde messengers that have been identified in specific cell types include dopamine, dynorphin, glutamate, GABA, nitric oxide, brain-derived neurotrophic factor (BDNF), and oxytocin (Drake et al, 1994; Kombian et al, 1997; Llano et al, 1991; Naggapan and Lu, 2005; Nugent et al, 2007; Pitler and Alger, 1992; Zilberter, 2000; Zilberter et al, 1999). Although postsynaptic, calcium-dependent fusion of vesicles is one common mechanism for the release of retrograde messengers, they can also released by non-vesicular mechanisms. For example, a widespread, extensively studied system for mediating retrograde synaptic signaling involves the postsynaptic release of endogenous cannabinoids, such as anandamide and 2-arachidonoylglycerol, which are produced upon demand by cleavage of phospholipids and are sensed by CB1 receptors on presynaptic terminals. The mechanisms of endocannabinoid release are unclear and may involve a transporter, which facilitates diffusion across the plasma membrane (Chevaleyre et al, 2006) Such retrograde signaling by postsynaptic release of endocannabinoids can be initiated by strong depolarization or activation of postsynaptic metabotropic receptors and has been shown to transiently suppress inhibitory and excitatory synapses in several brain regions (Chevaleyre et al, 2006). Importantly, in the striatum, there is a form of LTD (to be discussed below) that is triggered by the release of endocannabinoids.
Involvement of Glia in Short-Term Plasticity
There is growing realization that glia may be involved in some forms of short-term plasticity (Araque et al, 2001; Haydon, 2001). With their intimate association with synapses, astrocytes and perisynaptic Schwann cells are well positioned to regulate synapses. They have an established role in clearance of neurotransmitter and may participate in synaptic plasticity by controlling the speed and extent of such clearance (Bergles et al, 1999; Danbolt, 2001). This can in turn impact on the degree of postsynaptic receptor activation and desensitization. Another way that glia may be involved in synaptic plasticity is by sensing extracellular messengers and then releasing substances that in turn can directly affect synaptic efficacy (Araque et al, 2001; Haydon, 2001). For example, glia express many different neurotransmitter receptors (eg, glutamate receptors), which when activated result in the release of substances (eg, ATP) that can then act on presynaptic terminals to regulate neurotransmitter release.
Functions of Short-Term Synaptic Plasticity
Short-term synaptic plasticity was originally established as behaviorally important from studies of simple organisms such as Aplysia (Kandel and Tauc, 1965). In the mammalian brain, an important consequence of short-term synaptic plasticity is to influence the information processing function of synapses, enabling them to act as filters with a wide range of properties. For example, synapses with a low initial probability of release function as high-pass filters, since they will facilitate during high-frequency action potential bursts while low-frequency bursts will not be transmitted with the same efficacy. In contrast, synapses with a high initial probability of release function as low-pass filters, since they will depress during high-frequency bursts but will reliably relay low-frequency activity (Abbott and Regehr, 2004). The filtering characteristics of a synapse can be adjusted through modulation of the initial release probability. This most commonly occurs due to the release of neuromodulators that, via activation of presynaptic receptors, reduce the probability of release. This changes the filtering characteristics of the synapse, causing facilitation to become predominant over depression. In this way, presynaptic inhibition can convert a synapse from a low-pass to a high-pass filter.
LONG-TERM SYNAPTIC PLASTICIY
It is widely believed that experience of any sort modifies subsequent behavior at least in part through activity-dependent, long-lasting modifications of synaptic strength. The brain encodes external and internal events as complex, spatio-temporal patterns of activity in large ensembles of neurons that can be conceptualized as ‘neural circuits’. A key feature defining the behavior of any given neural circuit is the pattern of synaptic weights that connect the individual neurons that comprise the circuit. A corollary to this hypothesis is that new information is stored (ie, memories are generated) when activity in a circuit causes a long-lasting change in the pattern of synaptic weights. This idea was put forward over 100 years ago by the Spanish Nobel laureate Santiago Ramon y Cajal, and was further advanced in the late 1940s by Donald Hebb, who proposed that associative memories are formed in the brain by a process of synaptic modification that strengthens connections when presynaptic activity correlates with postsynaptic firing (Hebb, 1949). This proposed function for synaptic plasticity, forming a memory trace following the detection of two coincident events, suggests an appealing cellular basis for behavioral phenomena such as Pavlovian classical conditioning (Pavlov, 1927).
Experimental support for the very existence of such long-lasting, activity-dependent changes in synaptic strength was lacking until the early 1970s when Bliss and colleagues (Bliss and Gardner-Medwin, 1973; Bliss and Lomo, 1973) reported that repetitive activation of excitatory synapses in the hippocampus caused a potentiation of synaptic strength that could last for hours or even days. Over the last three decades, this phenomenon, eventually termed LTP, has been the object of intense investigation because it is widely believed that it provides an important key to understanding some of the cellular and molecular mechanisms by which memories are formed (Martin et al, 2000; Pastalkova et al, 2006; Whitlock et al, 2006).
Although still considered prototypic, it is now clear that hippocampal LTP is only one of several different forms of long-term synaptic plasticity that exist in specific circuits in the mammalian brain. Importantly, it is well established that most synapses that exhibit LTP also express one or more forms of LTD. Thus, a key concept is that synaptic strength at excitatory synapses is bidirectionally modifiable by different patterns of activity. Furthermore, it is now clear that the terms ‘LTP’ and ‘LTD’ describe a class of phenomena, the underlying mechanisms of which vary depending on the circuits in which they function.
Additional forms of synaptic plasticity more recently identified include homeostatic plasticity (Turrigiano and Nelson, 2004) and metaplasticity (Abraham and Bear, 1996). The major form of homeostatic plasticity is ‘synaptic scaling’, which describes phenomena whereby the strength of all synapses on a given cell are adjusted in response to prolonged changes in activity. Specifically, prolonged decreases in overall activity cause a net scaling up of total synaptic strengths while prolonged increases in activity cause the opposite, a scaling down of synaptic strengths. This form of plasticity operates on a much slower timescale than LTP or LTD and may be particularly important during the development of neural circuits. Metaplasticity refers to the effects that activity can have on the capacity of synapses to express long-term plasticity.
The most extensively studied and therefore prototypic forms of synaptic plasticity are the LTP and LTD observed in the CA1 region of the hippocampus (Figure 1), which are triggered by activation of N-methyl-D-aspartate (NMDA) receptors (NMDARs). We will therefore begin with a discussion of their underlying mechanisms. We will then proceed to reviewing some of the other prominent forms of LTP and LTD for which mechanisms have been most firmly established.

NMDAR-dependent LTP and LTD at hippocampal CA1 synapses. (a) Sample experiments illustrating LTP and LTD in the CA1 region of the hippocampus. Synaptic strength, defined as the initial slope of the field excitatory postsynaptic potential (fEPSP; normalized to baseline) is plotted as a function of time. Left panel demonstrates LTP elicited by high-frequency tetanic stimulation (100 Hz stimulation for 1 s; black arrowhead). Right panel illustrates LTD elicited by low-frequency stimulation (5 Hz stimulation for 3 min given twice with a 3 min interval; open arrow). Data traces were taken at the times indicated by the numbers on the graphs (scale bar: 0.5 mV; 10 ms) (courtesy of W Morishita). (b) A schematic diagram of the rodent hippocampal slice preparation, demonstrating the CA1 and CA3 regions as well as the dentate gyrus (DG). (SC=Schaffer collateral; MF=mossy fiber). Typical electrode placements for studying synaptic plasticity at Schaffer collateral synapses onto CA1 neurons are indicated (Stim=stimulating electrode; Rec=recording electrode). (c) Model of synaptic transmission at excitatory synapses. During basal synaptic transmission (left panel), synaptically released glutamate binds both the NMDA and AMPARs. Na+ flows through the AMPAR channel but not through the NMDAR channel because of the Mg2+ block of this channel. Depolarization of the postsynaptic cell (right) relieves the Mg2+ block of the NMDAR channel and allows both Na+ and Ca2+ to flow into the dendritic spine. The resultant increase in Ca2+ in the dendritic spine is necessary for triggering the subsequent events that drive synaptic plasticity.
NMDAR-Dependent LTP
No form of plasticity has generated more interest, or been more extensively studied than LTP in the CA1 region of the hippocampus. The excitement surrounding this phenomenon is due to compelling evidence from rodents, primates, and humans associating the hippocampus with a neural system involved in various forms of long-term memory (Martin et al, 2000; Zola-Morgan and Squire, 1993). Furthermore, several basic properties of LTP make it an attractive cellular mechanism for rapid information storage. Similar to memory, LTP can be generated rapidly and is strengthened and prolonged by repetition. It also exhibits cooperativity, associativity, and input specificity. (Nicoll et al, 1988). Cooperativity means that LTP can be induced by the coincident activation of a critical number of synapses. Associativity is the capacity to potentiate a weak input (a small number of synapses) when it is activated in association with a strong input (a larger number of synapses). As such, associativity is a cellular analogue of classical conditioning and is an implicit property of the so-called Hebbian synapse. Input specificity indicates that LTP is elicited only at activated synapses and not at adjacent, inactive synapses on the same postsynaptic cell. This feature dramatically increases the storage capacity of individual neurons since different synapses on the same cell can be involved in separate circuits encoding different bits of information.
A major technological advance in the study of synaptic plasticity was the development of the hippocampal slice preparation that made LTP accessible to rigorous experimental analysis (Figure 1b). Indeed, the bulk of our knowledge on the molecular mechanisms of LTP has been derived from studies of LTP at excitatory synapses on CA1 pyramidal neurons in hippocampal slices. Similar or identical forms of LTP have been observed at excitatory synapses throughout the brain. Thus, the conclusions drawn from the study of LTP in the hippocampal CA1 region are often applied to other brain regions.
Triggering NMDAR-dependent LTP
A major advance in the understanding of excitatory synaptic function and LTP was the demonstration that two major types of ionotropic glutamate receptors contribute to the postsynaptic response at glutamatergic synapses, α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptors (AMPARs) and NMDARs (Figure 1c). These receptors are often (although not always, as will be elaborated upon later) found colocalized on individual dendritic spines. The AMPAR has a channel that is permeable to monovalent cations (Na+ and K+), and activation of AMPARs provides most of the inward current that generates the excitatory synaptic response when the cell is close to its resting membrane potential. In contrast to AMPARs, the NMDAR exhibits a strong voltage dependence because of the block of its channel at negative membrane potentials by extracellular magnesium (Mayer et al, 1984; Nowak et al, 1984). As a result, NMDARs contribute little to the postsynaptic response during basal synaptic activity. However, when the cell is depolarized, magnesium dissociates from its binding site within the NMDAR channel, allowing ions to enter the cell. Importantly, unlike AMPAR channels, the NMDAR channel allows calcium as well as sodium to enter the postsynaptic dendritic spine (Figure 1c).
It is firmly established that the triggering (also termed the induction) of LTP in the CA1 region requires activation of NMDARs during strong postsynaptic depolarization leading to a increase in postsynaptic calcium concentration, which likely has to reach some critical threshold value to activate the biochemical processes necessary for LTP (Malenka, 1991; Malenka and Nicoll, 1993). Experimentally, this is normally achieved by applying high-frequency tetanic stimulation to the synapses or by use of a ‘pairing-protocol’ during which the postsynaptic cell is directly depolarized while low-frequency synaptic activation is sustained. An additional method for induction of LTP (as well as LTD) involves protocols that generate so-called ‘spike-time dependent plasticity’ (STDP; Dan and Poo, 2006; Markram et al, 1997). In studies of STDP, LTP is induced if afferent stimulation generates a synaptic response within a discrete time window prior to the firing of the postsynaptic cell.
Because its contribution to postsynaptic reponses requires both presynaptic release of glutamate and postsynaptic depolarization due to the simultaneous activation of a population of synapses, the NMDAR is often referred to as a ‘coincidence detector’. These properties of NMDARs also explain the basic properties of LTP. Cooperativity and associativity occur because of the requirement for multiple synapses to be activated simultaneously to generate adequate postsynaptic depolarization to remove the magnesium block of the NMDAR. Input specificity is due to the compartmentalized increase in calcium, which is limited to the postsynaptic dendritic spine and does not influence adjacent spines (Nicoll et al, 1988).
LTP signal transduction mechanisms
An extensive number of signal transduction molecules have been suggested to play a role in translating the calcium signal that is required to trigger LTP into the long-lasting increase in synaptic strength (Malenka and Bear, 2004; Sanes and Lichtman, 1999). However, only for a handful of these has compelling evidence of a mandatory role in LTP been presented. A major limitation of much of the literature on this topic stems from inadequate distinctions between molecules that are key components of the molecular machinery directly responsible for the triggering of LTP (‘mediators’), and those molecules which may modulate the ability to generate LTP, or play a permissive role (‘modulators’). Some basic criteria that can be suggested for defining the role of a protein as a mediator of LTP induction are: (1) blocking the activation of the molecule during LTP induction blocks LTP; and (2) activation of the molecule induces a potentiation of synaptic transmission, which occludes further synaptic induction of LTP.
Strong evidence indicates that calcium/calmodulin (CaM)-dependent protein kinase II (CaMKII) fulfills these requirements and is a key component of the molecular machinery for LTP. CaMKII undergoes autophosphorylation after the triggering of LTP (Barria et al, 1997; Fukunaga et al, 1995), and LTP induction was prevented both in knockout mice lacking a critical CaMKII subunit (Silva et al, 1992), and in knock-in animals in which endogenous CaMKII was replaced with a form lacking the autophosphorylation site (Giese et al, 1998). Furthermore, inhibition of CaMKII activity by directly loading postsynaptic cells with peptides that impair CaMKII function blocks LTP (Malenka et al, 1989; Malinow et al, 1989), whereas acutely increasing the postsynaptic concentration of active CaMKII increases synaptic strength and occludes LTP (Lledo et al, 1995; Pettit et al, 1994).
Several other kinases have been implicated in the triggering of LTP, but the experimental evidence supporting their role is not as well substantiated as that for CaMKII. Activation of the cyclic adenosine monophosphate-dependent protein kinase (PKA), perhaps by the activation of a calmodulin-dependent adenylyl cyclase, has been suggested to boost the activity of CaMKII indirectly by decreasing competing protein phosphatase activity (Blitzer et al, 1998; Lisman, 1989; Makhinson et al, 1999). This presumably happens by phosphorylation of inhibitor 1, an endogenous inhibitor of protein phosphatase 1 (PP1). The extracellular signal-regulated kinase (Erk)/mitogen-activated protein kinase (MAPK) pathway has also been suggested to be important for LTP, as well as some forms of learning and memory (Sweatt, 2004; Thomas and Huganir, 2004). In addition, Src kinase has been implicated in the enhancement of NMDAR function during LTP induction (Kalia et al, 2004). Finally, protein kinase C and in particular the atypical PKC isozyme, PKMζ, has received attention because this isozyme is rapidly expressed upon induction of LTP and recent studies have implicated PKMζ in the maintenance of the late phase of LTP both in hippocampal slices and in vivo (Hrabetova and Sacktor, 1996; Ling et al, 2002; Pastalkova et al, 2006; Serrano et al, 2005).
Obviously, it remains a very challenging task to be able to definitively identify the key intracellular signaling cascades responsible for the triggering of LTP. Recent technical advances in mass spectrometric techniques for the profiling of post-transcriptional modifications in mixed populations of proteins, as well as the practicality of RNAi approaches for knockdown of candidate proteins, should help further characterize the major LTP players and their dynamic interplay.
Expression mechanisms of LTP
In the past, a major point of contention was whether LTP was primarily expressed postsynaptically as a change in AMPAR properties or presynaptically, as a change in the probability of transmitter release. This latter expression mechanism garnered significant attention because it required the production of a retrograde messenger that was released by postsynaptic cells and acted on presynaptic terminals (for a thorough review of the evidence for either locus of expression, see Nicoll, 2003). To a large extent, this controversy has been resolved, with the emerging consensus that the major mechanism of expression of LTP at hippocampal CA1 synapses involves an increase in the numbers of AMPARs within the postsynaptic density, driven through activity-dependent changes in AMPAR trafficking (Bredt and Nicoll, 2003; Derkach et al, 2007; Malenka and Nicoll, 1999; Malinow and Malenka, 2002; Song and Huganir, 2002). A major contribution to ending the controversy was the proposal of the silent synapse hypothesis and the evidence supporting it (Durand et al, 1996; Isaac et al, 1995; Liao et al, 1995; Malenka and Nicoll, 1997). Silent synapses are synapses that contain only NMDARs with few or no AMPARs, such that at normal resting membrane potentials these synapses exhibit no detectable postsynaptic responses to synaptically released glutamate. The ‘unsilencing’ of these synapses during the induction of LTP is thought to occur through the incorporation of AMPARs into the postsynaptic plasma membrane. This hypothesis was quickly expanded to include synapses that already contained AMPARs and has led to a large investigative effort into the molecular mechanisms regulating the trafficking of AMPARs (Bredt and Nicoll, 2003; Collingridge et al, 2004; Malinow and Malenka, 2002; Sheng and Kim, 2002; Song and Huganir, 2002).
Most AMPARs in the central nervous system are tetramers comprised of four glutamate receptor subunits, GluR1–GluR4. Although these subunits are highly homologous, both the functional properties of AMPARs and their trafficking has been suggested to depend on their subunit composition (Collingridge et al, 2004; Derkach et al, 2007; Malinow and Malenka, 2002). In the adult hippocampus, two forms of AMPARs are thought to predominate: GluR1/GluR2 heteromers and GluR2/GluR3 heteromers (Wenthold et al, 1996). Based on clever overexpression studies, one prominent hypothesis suggests that subunit-specific rules govern the synaptic delivery of AMPARs (Hayashi et al, 2000; Passafaro et al, 2001; Shi et al, 2001). Specifically, it has been suggested that the insertion of GluR1-containing AMPARs into synapses is slow under basal conditions and is strongly stimulated by NMDAR activation, whereas insertion of GluR2/3 heteromeric receptors may occur constitutively on a much more rapid timescale.
Where do these new AMPARs come from and how is their trafficking regulated at a molecular level? Current evidence suggests that recycling endosomes in the dendrites contain a reserve pool of AMPARs that are mobilized during LTP via a process that requires the small GTP-binding protein, Rab11a (Park et al, 2004) (Figure 2). Surprisingly, AMPARs do not appear to be inserted directly into the postsynaptic density (PSD) but rather are exocytosed at perisynaptic sites. They then can laterally diffuse in the plasma membrane and be trapped within the PSD due to their interactions with so-called ‘slot proteins’, which function to trap AMPARs and greatly reduce their lateral mobility. Attractive candidates for these slot proteins are a family of proteins found in the PSD termed MAGUKs (for membrane-associated guanylate kinases). MAGUKS are defined by their multiple protein interaction domains, most importantly so-called PDZ domains. Prominent members of the MAGUK family of PSD proteins include PSD-95, SAP97, PSD-93, and SAP102 (Kim and Sheng, 2004; Montgomery et al, 2004). PSD-95 has received the most attention and its level appears to be particularly important for controlling the number of AMPARs at individual synapses as evidenced by the findings that overexpression of PSD-95 increases synaptic strength and occludes LTP (Ehrlich and Malinow, 2004; Stein et al, 2003), whereas knockdown of PSD-95 decreases surface expression of AMPARs and synaptic strength (Ehrlich et al, 2007; Schluter et al, 2006).

Model of AMPAR trafficking during LTP and LTD. In the basal state (depicted on top), receptors cycle between the postsynaptic membrane and intracellular compartments. This is achieved through lateral mobility of the receptors out of the synapse into endocytic zones, where they are endocytosed into early endosomes in a clathrin- and dynamin-dependent manner. Normally, the receptors are transferred to recycling endosomes and returned to the plasma membrane by exocytosis, followed by lateral movement into the synapse where they are retained through interaction with MAGUKs. Following induction of LTP, there is enhanced receptor exocytosis and stabilization at the synapse through a calcium-driven process that involves CAMKII and fusion of recycling endosomes mediated by Rab11a. Following the induction of LTD, enhanced endocytosis at extrasynaptic sites occurs in a process that is calcium-dependent and involves protein phosphatases, primarily calcineurin and protein phosphatases 1 (PP1). While in the basal state endocytosis is presumably balanced by receptor recycling, following LTD receptors are retained within the cell, and perhaps degraded.
The influence of PSD-95, as well as other MAGUKS (Elias et al, 2006; Futai et al, 2007; Schluter et al, 2006), on AMPARs was surprising given that PSD-95 was originally isolated because of its strong interaction with NMDARs (Kornau et al, 1995). Another major surprise was that AMPARs do not directly bind to MAGUKs but rather do so via a family of AMPAR auxiliary subunits termed transmembrane AMPAR regulatory proteins (TARPs). TARPs are required for the delivery of AMPARs to the plasma membrane at extrasynaptic sites (Chen et al, 2000; Nicoll et al, 2006) and also influence their biophysical properties (Tomita et al, 2005a). Importantly, they are also required for the synaptic localization of AMPARs due to their direct interaction with MAGUKs (Schnell et al, 2002).
The detailed molecular mechanisms by which activation of protein kinases such as CaMKII lead to the synaptic delivery of AMPARs remains to be determined. Phosphorylation of AMPAR subunits themselves by CaMKII does not appear critical (Esteban et al, 2003; Lee et al, 2003) although other phosphorylation sites, such as the PKA or the PKC sites on GluR1, may be important (Boehm et al, 2006; Esteban et al, 2003). On the other hand, there is reasonable evidence that phosphorylation of TARPs occurs following CAMKII activation (Tsui and Malenka, 2006) and that this may be critical for LTP (Tomita et al, 2005b). It is, however, highly likely that AMPARs and TARPs are just a component of a large complex of proteins, the phosphorylation of which will be required for LTP expression.
LTP also appears to involve a phosphorylation-driven increase in the single-channel conductance of AMPARs themselves (Benke et al, 1998; Soderling and Derkach, 2000). Indeed, CaMKII phosphorylates Ser831 in the intracellular C terminus of GluR1, resulting in a significant increase in single-channel conductance of homomeric GluR1 receptors (Derkach et al, 1999, 2007). However, one important caveat of this conclusion is that synaptic GluR1-containing AMPARs also contain GluR2, and in GluR1/2 heteromeric AMPARs, the enhanced conductance upon phosphorylation by CaMKII is absent (Oh and Derkach, 2005).
Although the evidence to date suggests a more prominent role for AMPAR exocytosis, the relative contribution of AMPAR trafficking and changes in the biophysical properties of AMPARs to the increase in synaptic strength during LTP are not well defined. One model for the successive events occurring during the first hour of LTP involves activation of calcium-dependent signal transduction pathways, notably CaMKII, resulting in phosphorylation of GluR1-containing receptors and an increase in their single-channel conductance. Roughly simultaneously, AMPARs are translocated into the PSD via exocytosis and lateral movement within the plasma membrane. Most investigators believe that the incorporation of AMPARs into the PSD is the more important change because it appears to be accompanied by structural changes in the dendritic spines and synapses themselves, an attractive mechanism for maintaining LTP (see below). There are also experimental findings consistent with rapid presynaptic changes during LTP, but the retrograde messenger that is responsible remains elusive, one prominent possibility being BDNF (Bramham and Messaoudi, 2005).
Maintaining LTP
Much of the work on NMDAR-dependent LTP has focused on the mechanisms responsible for its initial 30–60 min, to a large extent because of technical limitations in the duration over which stable electrophysiological recordings can be maintained. Nonetheless, the mechanisms that allow LTP to persist for hours, days, or even longer are of great importance. Like virtually all cell biological phenomena, the persistence of LTP is dependent upon new protein synthesis (Reymann and Frey, 2007). This so-called ‘late phase of LTP’ (defined as the potentiation present more than 1–2 h after LTP induction) is commonly assumed to depend upon local dendritic protein synthesis, which supplies needed components to the synapse (Sutton and Schuman, 2006), as well as transcription in the nucleus (Zhou et al, 2006). The signaling to the nucleus required for long-lasting LTP has been suggested to depend on a number of protein kinases including PKA, CaMKIV, and Erk-MAPK, which activate key transcription factors that may include cAMP response element-binding protein and immediate-early genes such as c-Fos and Zif268/Egr-1 (Thomas and Huganir, 2004). These transcriptional complexes presumably promote expression of effector genes that are required for maintaining the synaptic enhancement.
Several mRNAs can be found in dendrites, including those of the AMPARs themselves, CaMKII, Arc, and proteins which may function to regulate receptor trafficking (Grooms et al, 2006; Job and Eberwine, 2001; Ju et al, 2004; Mayford et al, 1996; Schuman et al, 2006; Steward and Schuman, 2001). The trafficking of some of these mRNAs and their local translation seems to be highly regulated by activity. Furthermore, other components of the translational machinery are found in or adjacent to dendritic spines and polyribosomes are recruited to spine heads following LTP induction (Bourne et al, 2007). Thus, there is accumulating evidence that the machinery to provide local, newly synthesized proteins to synapses is available.
An intriguing hypothesis is that during the synaptic activation to induce LTP a ‘synaptic tag’ is generated that functions to capture or sequester plasticity related proteins, which in turn are required to stabilize the increase in synaptic strength (Frey and Morris, 1997). However, little is known about the identity of the synaptic tag or the newly synthesized proteins that are required to maintain LTP, although it has been suggested that PKA, CAMKII, or PKMζ might function as the synaptic tag (Reymann and Frey, 2007; Sajikumar et al, 2005; Young et al, 2006).
A compelling possibility for a long-term maintenance mechanism of LTP is the structural remodeling of potentiated synapses (Luscher et al, 2000). Spines come in a variety of shapes and sizes, and can undergo rapid shape changes that are influenced by activity (Yuste and Bonhoeffer, 2001). Morphological changes which have been reported to accompany LTP include growth of new dendritic spines, enlargement of preexisiting spines and their PSDs, and the splitting of single PSDs and spines into two functional synapses (Abraham and Williams, 2003; Matsuzaki et al, 2004; Yuste and Bonhoeffer, 2001). An attractive model suggests that during LTP, recycling endosomes contribute AMPAR subunits to the synapse, as well as lipids and constituents which enlarge the synapse (Lisman and Zhabotinsky, 2001; Luscher et al, 2000; Park et al, 2004, 2006). At some later time point, there is likely a concomitant increase in the presynaptic active zone, the size of which always closely matches that of the PSD (Lisman and Harris, 1993). This presynaptic re-modeling must involve post- and presynaptic protein interactions with likely candidates being cell adhesion molecules such as the cadherins or neuroligin/neurexin interactions.
Summary
The current view of the mechanisms underlying this form of LTP can be summarized as follows (Figure 2). A large NMDAR-dependent increase in dendritic spine calcium concentrations leads to activation of intracellular signaling cascades involving a number of protein kinases, most importantly CaMKII. This leads to an increase in the single-channel conductance of synaptic AMPARs and more importantly, promotes the incorporation of additional AMPARs into the PSD. The new synaptic AMPARs are stabilized through their TARP-mediated interaction with PDZ domain-containing proteins such as PSD-95. In parallel, structural changes within the synapse occur, such that the size of the PSD and dendritic spine are increased. This in turn drives an increase in the size of the presynaptic active zone such that the potentiated synapses are ‘permanently’ enlarged. The maintenance of these changes for more than a few hours depends on de novo transcription as well as local dendritic protein synthesis, presumbably to provide the synapses with a supply of the critical proteins necessary for maintaining synaptic strength.
NMDAR-Dependent LTD
An important advance in the study of long-term synaptic plasticity was the establishment of an experimentally reproducible form of NMDAR-dependent LTD at excitatory synapses on hippocampal CA1 pyramidal cells (Dudek and Bear, 1992). This demonstrated that activity could bidirectionally control synaptic strength (Mulkey and Malenka, 1992), thus providing support for the idea that memories or experiences were encoded by the distribution of synaptic weights in neural circuits, not simply by LTP. Shortly thereafter, a similar form of LTD was observed at neocortical synapses (Kirkwood et al, 1993) suggesting that mechanistic studies in the hippocampal CA1 region could provide insights that applied to excitatory synapses throughout the brain. While that remains true to a large extent, we now know that in addition to NMDAR-dependent LTD, there are several other forms of LTD, which we will discuss in later sections. Moreover, similar to LTP, evidence is accumulating that LTD mechanisms may contribute to a large number of brain phenomena including experience-dependent development, learning and memory, addiction, and neurological disorders such as Alzheimer's disease and Parkinson's disease (Malenka and Bear, 2004; Brebner et al, 2005; Kreitzer and Malenka 2007; Hsieh et al, 2006).
Triggering NMDAR-dependent LTD
The typical protocol for eliciting LTD involves prolonged repetitive low-frequency stimulation (∼900 stimuli at 1 Hz; Dudek and Bear, 1992; Mulkey and Malenka, 1992), although the number of stimuli can be dramatically reduced and the frequency changed if the postsynaptic neuron is modestly depolarized (to ∼−50 mV), partially relieving the Mg2+ block of the NMDAR (Selig et al, 1995). NMDAR-dependent LTD can also be induced by correctly timing the activation of presynaptic axons and the postsynaptic neuron (STDP; (Dan and Poo, 2006).
Like LTP, LTD is input-specific and depends upon an NMDAR-dependent increase in postsynaptic calcium (Mulkey and Malenka, 1992). The predominant current hypothesis is that quantitative properties of the postsynaptic calcium signal within dendritic spines dictates whether LTP or LTD is triggered, with LTD requiring a modest increase in calcium (Cummings et al, 1996), whereas LTP requires a increase beyond some critical threshold value (Malenka and Nicoll, 1993). The temporal characteristics of the increase in calcium may also be important since changing the relative timing between pre- and postsynaptic activation by just a few tens of milliseconds can reverse the direction of synaptic modification (Dan and Poo, 2006). Differential buffering of postsynaptic calcium levels has been reported to enable a transition between LTP and LTD (Harney et al, 2006; Nishiyama et al, 2000). These findings demonstrate the thin line that lies between synaptic activity eliciting a potentiation vs a depression and provide support to the idea that both LTP and LTD are utilized in parallel for encoding experiences in vivo.
LTD signal transduction mechanisms
An influential hypothesis for the signal transduction pathway triggering LTD suggested that while LTP was due to the preferential activation of CaMKII, LTD involves activation of a calcium-dependent protein phosphatase cascade consisting of the calcium/calmodulin-dependent phosphatase calcineurin (also known as protein phosphatase 2B), PP1, and a phosphoprotein termed inhibitor-1 which functions to inhibit PP1 until it is dephosphorylated by calcineurin (Lisman, 1989). Consistent with this hypothesis, postsynaptic inhibition of these phosphatases prevents LTD (Kirkwood and Bear, 1994; Morishita et al, 2001; Mulkey et al, 1994, 1993), whereas directly loading CA1 pyramidal cells with PP1 enhances LTD (Morishita et al, 2001). Although signaling proteins other than phosphatases have been suggested to play key roles in LTD (Bolshakov et al, 2000; Dan and Poo, 2006; Palmer et al, 2005; Peineau et al, 2007), the hypothesis that LTP involves preferential activation of protein kinases while LTD involves activation of phosphatases remains predominant.
LTD has been found to correlate with dephosphorylation of postsynaptic PKC and PKA substrates without a detectable change in CaMKII substrate phosphorylation (Hrabetova and Sacktor, 1996; Kameyama et al, 1998; Lee et al, 2000, 1998. The evidence in support of a role for dephosphorylation of PKA substrates during LTD is particularly convincing, as postsynaptic inhibition of PKA, or its displacement from intracellular anchoring proteins, cause a rundown of synaptic transmission that occludes LTD. The mechanism underlying specific dephosphorylation of PKA substrates during LTD could involve the NMDAR-dependent recruitment of PP1 to synapses (Morishita et al, 2001) or the selective loss of PKA from synapses (Gomez et al, 2002; Snyder et al, 2005). Consistent with a role for PKA, LTD is associated with selective dephosphorylation of Ser845 on GluR1, a PKA substrate site (Lee et al, 2000). This dephosphorylation event may contribute to the expression of LTD, as it decreases the AMPAR open-channel probability (Banke et al, 2000), and mice with knock-in alanine substitution of both Ser845 and Ser831 (a CaMKII-substrate site) exhibit impaired NMDAR-dependent LTD (Lee et al, 2003).
Expression mechanisms of LTD
The demonstration of silent synapses and their conversion to functional synapses during LTP by the incorporation of AMPARs (Malenka and Nicoll, 1997) immediately gave credence to the idea that the opposite might happen during LTD, that is, the removal or endocytosis of AMPARs. Consistent with this idea, it was demonstrated that pharmacological manipulations of activity in cultured neurons (Carroll et al, 1999; Lissin et al, 1998) or application of glutamate receptor agonists (Beattie et al, 2000; Carroll et al, 1999; Ehlers, 2000; Heynen et al, 2000; Lin et al, 2000; Lissin et al, 1998) could cause a loss of AMPARs from synapses due to dynamin- and clathrin-dependent endocytosis (Carroll et al, 2001; Collingridge et al, 2004; Derkach et al, 2007; Malinow and Malenka, 2002). Furthermore, postsynaptic inhibition of dynamin activity impaired LTD in CA1 pyramidal cells in slices (Lee et al, 2002; Luscher et al, 1999; Morishita et al, 2005). Based in large part on these sorts of observations, the current leading hypothesis is that the expression mechanism of NMDAR-dependent LTD is due to activity-dependent endocytosis of synaptic AMPARs (Bredt and Nicoll, 2003; Collingridge et al, 2004; Derkach et al, 2007; Malenka and Bear, 2004; Malinow and Malenka, 2002) (Figure 2).
The precise molecular mechanisms by which endocytosis of AMPARs occurs during LTD is still not clear but one likely scenario is that it involves dissociation of AMPARs from their anchors within the PSD, followed by lateral movement to the edge of the PSD where they undergo clathrin- and dynamin-dependent endocytosis (Ashby et al, 2004; Blanpied et al, 2002; Groc et al, 2004). Consistent with a key role for protein phosphatases, the endocytosis of AMPARs is regulated by calcium-dependent dephosphorylation (Beattie et al, 2000; Carroll et al, 2001; Ehlers, 2000). Key substrates for facilitating endocytosis may include components of the endocytic machinery (Carroll et al, 2001; Lai et al, 1999; Slepnev et al, 1998) as well as AMPAR subunits themselves (Ehlers, 2000). Following endocytosis, AMPARs exhibit increased colocalization with the clathrin adaptor protein AP2 (Carroll et al, 1999), an interaction that may be promoted by the Ca2+-dependent adaptor protein hippocalcin (Palmer et al, 2005).
Consistent with a role for ‘slot proteins’ in the regulation of AMPAR surface levels, LTD has been reported to depend on PP1-dependent dephosphorylation of stargazin (Tomita et al, 2005b). This dephosphorylation might permit the dissociation of stargazin from PSD-95 and its diffusion out of the PSD, enabling LTD through loss of synaptic AMPARs (Ziff, 2007). Ubiquitination (Colledge et al, 2003) or depalmitoylation (El-Husseini et al, 2002) of PSD-95 has also been suggested to be critical for agonist-induced endocytosis of AMPARs but whether these events are required for synaptically induced LTD has not been determined. Additional proteins implicated in AMPAR endocytosis include the AMPAR interacting protein PICK1 (Hanley and Henley, 2005; Kim et al, 2001), the small GTPases Rap1 (Zhu et al, 2002, 2005) and Rab5 (Brown et al, 2005), and a protein termed cpg2 (Cottrell et al, 2004).
It has been suggested that particular AMPAR subunits, specifically GluR2, may be especially important for the endocytosis of AMPARs during LTD, in part because of its direct interaction with AP2 (Lee et al, 2002). One key finding consistent with this hypothesis is that peptides that selectively interfere with this interaction have been reported to inhibit LTD (Lee et al, 2002). However, AP2 can bind other AMPAR subunits (Kastning et al, 2007; Lee et al, 2002) and LTD is still observed in hippocampal slices obtained from mice lacking GluR2 as well as GluR2 and GluR3 (Jia et al, 1996; Meng et al, 2003). Thus, the importance of specific AMPAR subunits for LTD is not firmly established.
Summary
A current simplified view of the mechanisms underlying NMDAR-dependent LTD can be summarized as follows (Figure 2): a modest increase in postsynaptic calcium concentration within dendritic spines due to modest activation of NMDARs leads to preferential activation of protein phosphatases (as well as a few other key signaling proteins). This leads to the dissociation of AMPARs from their molecular scaffolds in the PSD and their lateral movement to endocytic zones on the periphery of the PSD, where they are endocytosed and potentially degraded. We have not discussed the maintenance of LTD because there is little work on this topic. There is evidence that LTD is accompanied by a shrinkage in the size of dendritic spines (Nagerl et al, 2004; Zhou et al, 2004) and that this may be due to the loss of AMPARs (Hsieh et al, 2006). Furthermore, similar to LTP, protein translation may be needed for the long-term stable expression of LTD (Pfeiffer and Huber, 2006). Thus, it is generally believed that the activity-dependent trafficking of AMPARs into and out of synapses during LTP and LTD, respectively, is the first critical step in the morphological growth or shrinkage of synapses and that these structural modifications are the mechanisms by which bidirectional changes in synaptic strength are maintained. Indeed, the size of individual synapses correlates closely with the number of AMPARs they contain (Matsuzaki et al, 2001; Nusser et al, 1998; Takumi et al, 1999).
Presynaptic LTP
Although the spotlight has been on NMDAR-dependent LTP for over 30 years, it is clear that a mechanistically distinct form of plasticity coexists in the hippocampus at the so-called mossy fiber synapses, the synapses between the axons of dentate gyrus granule cells (ie, mossy fibers) and the proximal apical dendrites of CA3 pyramidal cells (Nicoll and Malenka, 1995; Nicoll and Schmitz, 2005). Mossy fiber LTP (MF-LTP) is of increasing interest due to the fact that it appears to be a prototype for mechanistically similar forms of LTP found in several other brain regions including the thalamus at corticothalamic synapses (Castro-Alamancos and Calcagnotto, 1999), the cerebellum at parallel fiber-Purkinje cell synapses (Linden, 1997; Salin et al, 1996), and perhaps in the striatum at cortico-striatal synapses (Spencer and Murphy, 2002). Thus, like NMDAR-dependent LTP, this form of LTP may play multiple functional roles in the brain.
In contrast to NMDAR-dependent LTP, the triggering and expression of this form of LTP is thought to be solely or predominantly presynaptic, thus we refer to it as presynaptic LTP. While somewhat controversial, most experimental evidence suggests that presynaptic LTP is triggered by high-frequency tetanic stimulation, which causes a large, activity-dependent increase in calcium concentration within presynaptic axon terminals (Nicoll and Malenka, 1995; Nicoll and Schmitz, 2005; Zalutsky and Nicoll, 1990). Presynaptic voltage-dependent calcium channels are the critical source of the calcium increase, although the triggering of this LTP at MF synapses can be facilitated by the activation of presynaptic kainate receptors, particularly GluR6 (Lauri et al, 2001; Schmitz et al, 2003). Results from pharmacological manipulations and knockout mice are all consistent with the hypothesis that the increase in presynaptic calcium activates a calcium/calmodulin-dependent adenylyl cyclase. This leads to a increase in presynaptic cAMP and activation of PKA, which phosphorylates critical presynaptic substrates to cause a long-lasting enhancement in transmitter release (Nicoll and Malenka, 1995; Nicoll and Schmitz, 2005). Although this sequence of events is consistent with most of the results from all of the synapses that have been reported to express presynaptic LTP, surprisingly, a very different postsynaptic induction mechanism has been proposed to occur at MF synapses. This involves trans-synaptic interactions between postsynaptic EphB receptor tyrosine kinases and presynaptic B-Ephrin ligands leading, via an unknown mechanism, to a long-lasting enhancement of transmitter release (Armstrong et al, 2006; Contractor et al, 2002).
Despite some controversy concerning the mechanism underlying the induction of presynaptic LTP at MF synapses, there is general agreement that its expression is due to an increase in neurotransmitter release. One mechanism for mediating a long-lasting increase in transmitter release is to enhance the calcium influx into the presynaptic terminal when it is invaded by an action potential; for example via a long-lasting modulation of presynaptic calcium channels. However, direct imaging of the action potential-dependent increase in presynaptic calcium in MF boutons indicates that no change occurs following the triggering of MF-LTP (Kamiya et al, 2002; Regehr and Tank, 1991; Reid et al, 2004).
The other major mechanism for enhancing transmitter release is via some modulation of the machinery responsible for synaptic vesicle exocytosis. In this context, the focus has been on presynaptic proteins known to be substrates for PKA. Because presynaptic terminals are relatively inaccessible, an important approach has been examining knockout mice lacking specific presynaptic proteins. Mice lacking synapsins exhibited normal MF-LTP (Spillane et al, 1995) indicating that these prominent PKA substrates were not necessary. In contrast, knockout animals lacking the synaptic vesicle protein Rab3A, a small GTPase (Castillo et al, 1997; Lonart et al, 1998), or its binding partner, the active zone protein Rim1α (Castillo et al, 2002; Powell et al, 2004) lack both MF-LTP as well as presynaptic LTP in the cerebellum. Rim1α is a substrate of PKA and thus these findings suggest that presynaptic LTP may require a GTP-dependent interaction between Rab3a and Rim1α at the interface of synaptic vesicles and the active zone. Indeed, using cultures of cerebellar neurons in which presynaptic LTP can be elicited, the critical residue phosphorylated by PKA in Rim1α has been identified (Lonart et al, 2003). One important caveat to this hypothesis, however, is that the presynaptic enhancement of synaptic transmission seen upon application of the adenylyl cyclase activator forskolin is unaltered in knockout mice lacking Rab3a or Rim1α. This provides a complication for the simple model implicating Rim1α and Rab3a as the crucial mediators of the enhanced transmission caused by presynaptic activation of PKA.
Metabotropic Glutamate Receptor-Dependent LTD
Under the appropriate experimental conditions, low-frequency stimulation of the Schaffer collateral/commissural inputs to CA1 pyramidal cells can trigger, in addition to NMDAR-dependent LTD, a mechanistically distinct form of LTD, that is dependent upon activation of metabotropic glutamate receptors (mGluRs; mGLUR LTD) (Anwyl, 2006; Bolshakov et al, 2000; Malenka and Bear, 2004; Oliet et al, 1997). Various forms of mGluR LTD have been observed in many brain regions, most notably at the parallel fiber to Purkinje cell synapse (Ito, 1989; Linden and Connor, 1995). This so-called cerebellar LTD is elicited when parallel fibers are activated coincident with climbing fiber activation and has been suggested to be particularly important for certain forms of motor learning.
Typically, the induction of mGluR LTD in the CA1 region of the hippocampus requires bursts of afferent stimulation or paired-pulse stimulation at modest frequencies. Presumably, this increases the likelihood of activating the extrasynaptic group I mGluRs that are required to trigger this form of plasticity. mGluR LTD can also be elicited by bath application of group I mGluR agonists such as DHPG (Bellone and Luscher, 2005; Huber et al, 2000, 2001). Group I mGluRs (mGluR1 and mGluR5) are defined by their capacity to stimulate phosphoinositide hydrolysis and thereby produce diacylglycerol and inositol triphosphate. Although mGluR1 is the principal receptor implicated in the induction of mGluR LTD in the ventral tegmental area (VTA), cerebellum, and neostriatum, mGluR5 has been implicated in the hippocampus, cortex, and nucleus accumbens (Anwyl, 2006).
The intracellular signaling pathways responsible for mGluR LTD have not been extensively examined. PKC appears to be required for mGluR LTD in the cerebellum (De Zeeuw et al, 1998; Linden and Connor, 1991) and the VTA (Bellone and Luscher, 2005), but perhaps not in the hippocampus (Huang and Hsu, 2006). Other signaling proteins suggested to be important for mGluR LTD include the MAPKs p38, Erk, and Jnk (Gallagher et al, 2004; Li et al, 2007; Rush et al, 2002), tyrosine phosphatases (Huang and Hsu, 2006; Moult et al, 2006), and phosphatidylinositol 3-kinase (Hou and Klann, 2004) but how these various enzymes might lead to a long-lasting decrease in synaptic strength is unknown.
Similar to NMDAR-dependent LTD, mGluR LTD in the hippocampus and cerebellum appears to involve clathrin-dependent endocytosis of AMPARs (Snyder et al, 2001; Wang and Linden, 2000; Xiao et al, 2001). For cerebellar mGluR LTD, a large body of work is consistent with a specific molecular model in which mGluR activation due to parallel fiber stimulation combined with calcium influx due to climbing fiber activation results in activation of PKCα and its targeting to synapses via the protein PICK1 (Jorntell and Hansel, 2006; Perez et al, 2001; Steinberg et al, 2006). PKCα then phosphorylates Ser880 on GluR2 causing GluR2 to dissociate from the AMPAR-binding proteins GRIP/ABP and bind to PICK1. This then allows lateral diffusion of the AMPARs and ultimately internalization (Chung et al, 2003; Steinberg et al, 2006; Xia et al, 2000). This cascade of events is significantly different than those believed to occur during NMDAR-dependent LTD, pointing out that the molecular mechanisms underlying apparently similar forms of plasticity may be cell type-specific. Indeed, mGluR LTD at excitatory synapses onto dopamine cells in the VTA has been suggested to involve a selective loss of AMPARs that lack the GluR2 subunit (Bellone and Luscher, 2005, 2006), rather than those that contain GluR2.
Another important feature of mGluR LTD, at least in the hippocampus, is that it appears to be dependent on mGluR-triggered protein synthesis (Hou and Klann, 2004; Huber et al, 2000, 2001; Snyder et al, 2001). This link between protein synthesis and mGluR LTD gained considerable attention following the report that mice lacking the gene encoding the fragile X mental retardation protein (FMRP) exhibited enhanced mGluR LTD in both the hippocampus (Huber et al, 2002) and the cerebellum (Koekkoek et al, 2005). These mice serve as a model for the Fragile X syndrome in humans, an X-linked form of mental retardation, and exhibit several features of the human disorder (Bear et al, 2004). Surprisingly, the enhanced mGluR LTD observed in the FMRP knockout mice is independent of protein synthesis and proteasomal degradation (Hou et al, 2006; Nosyreva and Huber, 2006), cellular processes that appear crucial for mGluR LTD in wild-type mice. Nevertheless, this potential link between an mGluR-triggered form of synaptic plasticity and Fragile X syndrome has focused attention on mGluR antagonists as possible therapeutic agents for this and other developemental disorders (Bear et al, 2004).
There are also results consistent with a presynaptic locus of expression for mGluR LTD (Anwyl, 2006; Nosyreva and Huber, 2005; Zakharenko et al, 2002). As these forms of mGluR LTD are still triggered postsynaptically, they must involve a retrograde messenger, one leading candidate being 12-lipoxygenase metabolites of arachidonic acid (Feinmark et al, 2003). In an attempt to reconcile the observations of a presynaptic locus of expression of mGluR LTD with the bulk of the literature describing a postsynaptic locus of expression, a developmental shift in the mechanisms of mGluR LTD has been proposed (Nosyreva and Huber, 2005). Specifically, it has been suggested that in slices prepared from neonatal (P8–P15) animals, mGluR-LTD is independent of protein synthesis and is due to a presynaptic modification, whereas in older animals (P21–P35) mGluR LTD is mediated by a protein synthesis-dependent reduction in the postsynaptic levels of AMPARs.
Endocannabinoid-Mediated LTD
A major advance over the last decade has been the demonstration of the existence of endogenous cannabinoids (termed endocannabinoids) and their roles in modulating synaptic function (Chevaleyre et al, 2006; Freund et al, 2003; Kreitzer and Regehr, 2002; Piomelli, 2003; Wilson and Nicoll, 2002). It is now well established that in a number of brain regions endocannabinoids are retrograde messengers that are released by postsynaptic cells in response to strong depolarization and/or activation of G-protein-coupled receptors (eg, mGluRs and muscarinic receptors) and function to transiently inhibit transmitter release (for ∼0.5–1 min) at either excitatory or inhibitory synpases via activation of presynaptic CB1 receptors.
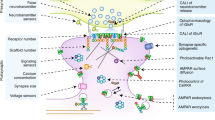
Much of the mechanistic work on the transient synaptic effects of endocannabinioids was performed in the hippocampus and cerebellum, synapses that are often thought to be prototypic of synapses throughout the brain. It was therefore surprising when a form of LTD that required endocannabinoids, (so-called endocannabinoid-mediated LTD; eCB-LTD) was observed in the glutamatergic synapses onto medium spiny neurons in the striatum as well as at synapses between layer V pyramidal neurons (Gerdeman et al, 2002; Robbe et al, 2002a; Sjostrom et al, 2003). In the hippocampus, in contrast, endocannabinoids mediate a form of LTD at inhibitory, but not excitatory, synapses (Chevaleyre and Castillo, 2004). In the dorsal striatum, eCB-LTD requires postsynaptic activation of group I mGluRs, L-type calcium channels and D2 dopamine receptors (Choi and Lovinger, 1997; Kreitzer and Malenka, 2005, 2007; Pisani et al, 2005; Sung et al, 2001; Tang et al, 2001) (Figure 3). This leads to the production most likely of anandamide (Ade and Lovinger, 2007), which activates presynaptic CB1 receptors. However, prolonged activation of these CB1 receptors alone is not sufficient to elicit eCB-LTD; concomitant presynaptic activity is also required, a requirement that likely accounts for the input specificity of this form of plasticity (Singla et al, 2007). Another important feature of eCB-LTD in the dorsal striatum is that it appears to be restricted to the medium spiny neurons that primarily express D2 dopamine receptors and is not present at excitatory synapses on cells expressing D1 receptors (Kreitzer and Malenka, 2007; but see Wang et al, 2006). This cell-restricted expression of eCB-LTD may have important functional implications, a topic to be discussed below.

Model of eCB-LTD at excitatory synapses onto medium spiny neurons in the striatum. Activation of postsynaptic type I mGluRs, along with coincident subthreshold depolarization of medium spiny neurons sufficient to activate L-type voltage-sensitive calcium channels (VSCCs), stimulates the postsynaptic synthesis and release of endocannabinoids. What enzyme generates the endocannabinoids is not known; one candidate is PLCβ. Co-activation of postsynaptic dopamine D2-type receptors (D2R) enhances endocannabinoid production and the subsequent induction of presynaptic LTD, mediated through activation of presynaptic CB1 receptors (CB1R). (From Kreitzer and Malenka, 2005.)
In the nucleus accumbens (NAc), eCB-LTD also requires postsynaptic activation of group I mGluRs and is modulated by drugs of abuse (Fourgeaud et al, 2004; Mato et al, 2004; Robbe et al, 2002b). In the cortex, eCB-LTD can be elicited by a spike-timing-dependent protocol (Sjostrom et al, 2003) but does not depend on postsynaptic activation of mGluRs and surprisingly, requires coincident activation of presynaptic NMDARs. LTD in the cerebellum was also reported to depend on retrograde endocannabinoid signaling (Safo and Regehr, 2005), but the mechanistic basis for this observation is still unclear, since, as discussed above, cerebellar LTD is expressed postsynaptically.
Metaplasticity
Metaplasticity refers to a higher-order form of synaptic plasticity in which synaptic activity, which by itself does not directly affect synaptic efficacy, leads to a persistent change in the direction or magnitude of subsequent activity-dependent synaptic plasticity. In other words, metaplasticity is the ‘plasticity of plasticity’ (Abraham and Bear, 1996; Bear et al, 1987). The best-studied examples of metaplasticity are those in which prior activity shifts the threshold for LTP and LTD induction. For example, in the hippocampus, repetitive activation of NMDARs in a manner that does not elicit LTP or LTD can nonetheless elicit a rapid shift in plasticity thresholds such that LTP becomes difficult to elicit and LTD induction is favored (Huang et al, 1992; Wang and Wagner, 1999). A potential functional role for metaplasticity has been demonstrated by modifying the level of activity during the development of the visual cortex in vivo (Philpot et al, 2001, 2003, 2007). This causes shifts in the thresholds for LTP and LTD in cells in visual cortex presumably due to changes in the stoichiometry of synaptic NMDARs. As discussed below, such changes in synaptic plasticity may importantly contribute to the experience-dependent plasticity of ocular dominance following manipulation of the visual environment.
Synaptic Scaling: A Form of Homeostatic Plasticity
Theoretically, without additional stabilizing mechanisms, activity-dependent forms of plasticity such as LTP and LTD could drive neural circuit activity towards epileptogenic excitation or complete quiescence. Synaptic scaling is considered a form of homeostatic plasticity that counters potentially maladaptive effects of long-term synapse-specific plasticity by globally affecting the transmission through all synapses on a given neuron (Turrigiano and Nelson, 2004). In terms of its basic properties and underlying mechanisms, this form of synaptic plasticity contrasts dramatically with the forms of LTP and LTD we have discussed thus far.
Synaptic scaling occurs when network activity is dramatically decreased or increased for prolonged (>∼12–24 h) periods of time. Decreased activity (due to blockade of synaptic transmission or spiking) causes an increase in the strength of all excitatory synapses onto excitatory neurons, whereas increased activity (generally induced by partially blocking inhibitory synapses) reduces the strength of all excitatory synapses (Turrigiano et al, 1998). Importantly, the relative strengths of individual synpases appear to be maintained even though global synaptic input is significantly altered.
Relatively little is known about the molecular mechanisms underlying synaptic scaling other than it involves changes in the number of AMPARs (and NMDARs) at individual synapses (Perez-Otano and Ehlers, 2005; Turrigiano and Nelson, 2004; Watt et al, 2000) and likely presynaptic changes as well (Burrone and Murthy, 2003; Burrone et al, 2002). Recently, evidence has been presented supporting a role for secreted factors in the induction of homeostatic plasticity, suggesting that key triggers for this form of plasticity may not be cell autonomous. Specifically, secretion of the proinflammatory cytokine tumor necrosis factor-α from glial cells appears to be necessary for the increase in the level of synaptic AMPARs caused by extended periods of activity blockade (Beattie et al, 2002; Stellwagen et al, 2005; Stellwagen and Malenka, 2006). In addition, there is evidence suggesting a role for secreted BDNF in driving the opposite form of synaptic scaling; the decrease in synaptic strengths caused by extended periods of increased network acitivity (Rutherford et al, 1998; Turrigiano, 2006).
FUNCTIONAL ROLES OF LTP AND LTD
Although LTP and LTD are prime candidate mechanisms underlying many different forms of experience-dependent plasticity, it is important to remember that they are experimental phenomena used to examine how different patterns of activity can elicit bidirectional control over synaptic strength. Establishing a causal connection between a specific form of synaptic plasticity and the behavioral consequences of specific experiences remains a daunting task. Nevertheless, over the last decade, significant advances have been made in connecting synaptic plasticity to a number of different types of adaptive experience-dependent plasticity. Furthermore, it has become increasingly clear that understanding the mechanisms of synaptic plasticity may provide important insights into the pathophysiology of a variety of neuropsychiatric disorders and also point the way toward novel therapeutic approaches. In the following sections, we will briefly provide examples that demonstrate that LTP and LTD do occur in vivo in response to experience and may play a causal role in mediating the consequences of experience.
Hippocampal-Dependent Plasticity and Learning
Given that LTP was first described in the hippocampus, a structure well established to be critically important for declarative memory (Squire et al, 2004), it is not surprising that over the last three decades there has been a major effort aimed at demonstrating a role for hippocampal LTP in encoding new memories (Martin et al, 2000; Morris, 2006). Correlations have been observed between defective hippocampal synaptic plasticity and defective hippocampal-dependent memory tasks upon perturbation of a number of proteins which function in synaptic plasticity, either pharmacologically, or through gene knockout (Lynch, 2004; Martin et al, 2000; Morris, 2006). For example, rodents in which NMDAR antagonists were infused into the hippocampus (Morris and Frey, 1997), as well as mice lacking expression of the NMDAR subunit NR1 in the forebrain (Tsien et al, 1996), are defective both in LTP and certain types of spatial learning. Furthermore, mice in which the specific NMDAR subunit NR2B was overexpressed to enhance NMDAR function were reported to display enhanced LTP and enhanced spatial learning (Tang et al, 1999). Recently, more compelling evidence for a role of synaptic plasticity in hippocampal-dependent learning has been presented. During an inhibitory avoidance task, LTP could be recorded in vivo in a subset of hippocampal CA1 pyramidal cells (Whitlock et al, 2006). This demonstrated that the patterns of activity generated during a real learning task were sufficient to elicit LTP. Perhaps even more convincing was the demonstration that in vivo infusion of a PKMζ inhibitor into the hippocampus abolished the maintenance of LTP and simultaneously the storage of a long-lasting spatial memory (Pastalkova et al, 2006). These findings strongly suggest that maintained LTP was required for the engram that stored the key spatial information.
In addition to its role as a key component of the mechanisms underlying the encoding of declarative memories, hippocampal NMDAR-dependent LTP (as well as LTD) may provide important insights into the pathophysiology and potential treatment of major mental illnesses. For example, a leading hypothesis for the pathophysiology of schizophrenia posits a dysfunction in glutamatergic synapses, in particular a hypofunction of NMDARs (Coyle and Tsai, 2004; Javitt, 2006; McCullumsmith et al, 2004; Tamminga, 1998). Thus, understanding the signaling events downstream of NMDAR activation may provide important insights into this devastating disease. A role for dysfunctions in LTP and/or LTD mechanisms as possible contributors to schizophrenia is also attractive in that schizophrenia likely involves neurodevelopmental abnormalities (Lewis and Levitt, 2002) and these phenomena play an important role in the early postnatal development of neural circuitry (see below). Indeed, genetic variation in calcineurin, which is thought to play an important role in NMDAR-dependent LTD, has been associated with schizophrenia (Gerber et al, 2003).
Another example of the potential importance of studying LTP and LTD comes from investigation into the therapeutic mechanisms of drugs used to treat bipolar disorder. Drugs such as lithium, valproate, and lamotrigine have been reported to have significant effects on the phosphorylation of AMPAR subunits and affect their surface expression (Du et al, 2003, 2004, 2007; Gray et al, 2003). These findings suggest that these drugs may somehow tap into the same mechanisms that have evolved to generate LTP and LTD and also point to novel approaches for the development of new therapeutic agents that may prove efficacious for treatment of this illness.
Experience-Dependent Plasticity in the Developing Sensory Cortex
Sensory receptive fields in the cortex are modified by early postnatal experience and the link between synaptic plasticity and these forms of experience-dependent plasticity in sensory systems is becoming increasingly established (Foeller and Feldman, 2004; Karmarkar and Dan, 2006; Malenka and Bear, 2004). For example, a strong connection between synaptic plasticity and experience-dependent plasticity has been established in the visual system during the shift in ocular dominance caused by monocular deprivation (MD) (Foeller and Feldman, 2004; Karmarkar and Dan, 2006; Malenka and Bear, 2004). MD induces biochemical changes in AMPAR subunits in visual cortex that appear identical to those elicited during NMDAR-dependent LTD (Heynen et al, 2003) and visual cortical slices obtained from monocularly deprived animals show greatly reduced LTD (Crozier et al, 2007; Heynen et al, 2003), suggesting that LTD was elicited in vivo. Furthermore, in vivo recordings demonstrated that MD caused a rapid decrease in the visually evoked potential (VEP) from the deprived eye and a slower enhancement of the VEP from the open eye (Frenkel and Bear, 2004). Importantly, completely blocking activity in the deprived eye prevented the depression of the VEP demonstrating that this depression required retinal activity and, like LTD, was therefore activity-dependent (Frenkel and Bear, 2004).
Similar results have been obtained in somatosensory barrel cortex in that sensory deprivation by whisker trimming or plucking causes a weakening of synaptic responses in layer 2/3 cells and an occlusion of LTD (Allen et al, 2003). This appears to be due to alterations in the patterns of pre- and postsynaptic spiking in vivo in a manner that is ideal for generating spike-timing-dependent LTD (Celikel et al, 2004). LTP mechanisms, on the other hand, appear to be important for the strengthening of synapses in developing barrel cortex due to early postnatal experience. In vivo experience drives recombinant GluR1 into barrel cortex synapses in a manner similar to that which occurs during NMDAR-dependent LTP, whereas expression of a short peptide that inhibits delivery of endogenous AMPARs blocks the experience-dependent increase in synaptic strength (Takahashi et al, 2003) as does a dominant-negative form of the synaptic scaffold protein PSD-95 (Ehrlich and Malinow, 2004). Thus, as predicted by theoretical considerations (Bienenstock et al, 1982; Stent, 1973), LTP and LTD mechanisms appear to be critically involved in early neural circuit development and it is not difficult to imagine how disruption in these mechanisms might contribute to a host of neurodevelopemental disorders such as autism (Geschwind and Levitt, 2007; Rubenstein and Merzenich, 2003).
Synaptic Plasticity and Fear Conditioning
Pavlovian fear conditioning is a form of associative memory that depends on the amygdala for its induction and maintenance (Sigurdsson et al, 2007). It occurs when a neutral stimulus (such as a tone) is temporally paired with a strong noxious stimulus (such as an electric shock) creating a memory trace, the consequences of which are that the neutral stimulus elicits the learned fear response. Considerable evidence is consistent with the hypothesis that LTP at sensory inputs to the lateral nucleus of the amygdala is necessary and perhaps sufficient for establishing this engram (Sigurdsson et al, 2007). NMDAR-dependent LTP can be induced at cortical and thalamic inputs into the lateral amygdala, both in vitro and in vivo. Importantly, fear conditioning induces synaptic potentiation at these synapses, and this increase in synaptic strength occludes further induction of LTP (McKernan and Shinnick-Gallagher, 1997; Rogan et al, 1997; Tsvetkov et al, 2002). Like NMDAR-dependent LTP in the hippocampus, fear conditioning has also been shown to lead to the insertion of new AMPARs at thalamic input synapses onto lateral amygdala neurons (Rumpel et al, 2005) and perhaps most convincingly, expression of a peptide that prevents the LTP-induced incorporation of AMPARs at synapses abolished acquisition of this form of associative memory (Rumpel et al, 2005).
NMDARs in the amgydala are also involved in the extinction of learned fear, which can be conceptualized as a different form of learning (Myers and Davis, 2002). Furthermore, acute treatment with D-cycloserine, a partial agonist of NMDARs, enhances the learning processes that are responsible for fear extinction via actions in the amygdala (Walker et al, 2002). On the basis of these observations, clinical trials have been initiated using D-cycloserine in combination with behavioral therapy to enhance the extinction of fear in phobic patients (Hofmann et al, 2006; Otto et al, 2007; Ressler et al, 2004). Results to date suggest that the administration of D-cycloserine either before or shortly after exposure to fearful cues does in fact enhance the extinction of the anxiety previously associated with specific cues. Thus, the study of the neural substrates of learned fear and its extinction is a compelling example of how research on the mechanisms of synaptic plasticity has directly led to a potential novel treatment for common psychiatric disorders.
Synaptic Plasticity in Dopamine Circuits
The defining characteristic of drug addiction is persistent and compulsive seeking and ingestion of drugs despite adverse consequences. Over the last decade, a leading hypothesis has been that an important neural substrate of addiction, in particular relapse, is long-term associative memory processes occurring in several neural circuits that receive input from midbrain dopamine neurons (Everitt and Robbins, 2005; Hyman et al, 2006). In other words, it is thought that addictive drugs can usurp the normal adaptive mechanisms underlying reward-based learning.
The most well-established key site of action of addictive drugs is the mesolimbic dopamine system consisting of the VTA and the NAc. Excitatory synaptic transmission in these structures is critical for mediating several forms of long-lasting, drug-induced behavioral plasticity (Everitt and Wolf, 2002; Hyman and Malenka, 2001). It, therefore, has been reasonable to hypothesize that plasticity at these synapses plays an important role in mediating some of the behavioral consequences of exposure to drugs of abuse (Wolf, 1998).
Indeed, it is now established that various forms of LTP and LTD can be elicited at excitatory synapses in the VTA and NAc (Gerdeman et al, 2003; Kauer, 2004; Kelley, 2004; Thomas and Malenka, 2003). Furthermore, administration of a single dose of several different classes of drugs of abuse causes a significant increase in synaptic strength at excitatory synapses onto dopamine cells in the VTA (Faleiro et al, 2004; Saal et al, 2003; Ungless et al, 2001). This increase shares mechanisms with LTP in the VTA and appears to involve upregulation of AMPARs (Ungless et al, 2001). This drug-induced ‘LTP’ appears to play a functional role in triggering or mediating some drug-induced behavioral adaptations, as both conditioned place preference and behavioral sensitization are blocked by injection of glutamate receptor antagonists into the VTA (Harris and Aston-Jones, 2003; Vanderschuren and Kalivas, 2000). In addition, conditioned place preference, as well as the synaptic potentiation that is observed following cocaine administration, are impaired in GluR1 knockout mice (Dong et al, 2004), with the caveat that these mice still exhibit robust behavioral sensitization in response to repeated exposure to psychostimulants (Dong et al, 2004; Vekovischeva et al, 2001).
While drugs of abuse have been observed to induce LTP in the VTA, LTD has been observed in the NAc following chronic in vivo cocaine administration (Thomas et al, 2001). Consistent with a functional role for this LTD in addiction, injection of glutamate receptor antagonists into the NAc abolishes the expression of behavioral sensitization (Kelley, 2004). Furthermore, amphetamine-induced behavioral sensitization and LTD in the NAc were both abolished by injection into the NAc of inhibitors of AMPAR endocytosis (Brebner et al, 2005). Interestingly, overexpression of GluR1 in the NAc (mimicking an ‘LTP’-like condition) facilitates the extinction of cocaine-seeking (Sutton et al, 2003) and even makes cocaine aversive in a conditioned place preference assay (Kelz et al, 1999). Recently, it has also been reported that cocaine self-administration abolishes the ability to induce LTD in the core of the NAc after prolonged (21 days) abstinence (Martin et al, 2006) and in vivo administration of tetrahydrocannabinol (THC) or cocaine impairs the generation of eCB-LTD (Fourgeaud et al, 2004; Mato et al, 2004; Robbe et al, 2002b). Thus, drugs of abuse may elicit certain forms of synaptic plasticity in specific circuits while simultaneously impairing plasticity in other circuits.
Synaptic plasticity in the dorsal striatum has also been correlated with certain learned behaviors, in particular in motor control (Gubellini et al, 2004; Pisani et al, 2005). In this context, it is important to note that two independent striatal circuits are thought to exist, the so-called ‘direct’ and ‘indirect’ pathways, which subserve different functions in motor control (Bolam et al, 2000). Indirect pathway medium spiny neurons (MSNs) project to the lateral globus pallidus and primarily express D2 dopamine receptors while direct pathway MSNs project to the substantia nigra and express D1 dopamine receptors. As mentioned in the section on eCB-LTD, this form of plasticity is primarily restricted to the indirect pathway MSNs and is absent in Parkinson's disease models (Kreitzer and Malenka, 2007). However, in slices from dopamine-depleted animals, indirect pathway eCB-LTD can be rescued by a D2 receptor agonist or pharmacological inhibitors of endocannabinoid degradation. Remarkably, administration of these drugs together in vivo dramatically reduces Parkinsonian motor deficits suggesting that endocannabinoid-mediated depression of indirect pathway synapses has a critical role in motor control and may be a valuable target for therapy of striatal-based brain disorders (Kreitzer and Malenka, 2007). This works also points out the potential power of examining the mechanisms of synaptic plasticity and placing them in the context of the neural circuits in which they are found.
Future Directions
We have attempted to briefly review the enormous field of synaptic plasticity research in a concise and accessible manner so that by the end of this article, readers will have a reasonably up to date knowledge of current thinking about the mechanisms underlying the major forms of synaptic plasticity in the mammalian brain and a sense of what their in vivo functions might be. By necessity we have had to leave out important topics such as how synaptic activity modulates NMDAR-mediated synaptic responses (eg, Morishita et al, 2005), inhibitory synaptic responses (Chevaleyre et al, 2006), or the intrinsic excitability of neurons (Xu and Kang, 2005; Zhang and Linden, 2003), all of which will have profound effects on neural circuit function. Furthermore, several compelling examples that correlate long-term synaptic plasticity with experience-dependent modifications in behavior have been left out, such as the role of cerebellar LTD in motor learning (Boyden et al, 2004; Ito, 2002; Jorntell and Hansel, 2006).
This review should also make clear that while extensive progress has been made, much remains unknown. Even for the most established forms of plasticity, NMDAR-dependent LTP and LTD in the CA1 region of the hippocampus, we still know little about the details of the signal transduction pathways triggering these forms of plasticity and which specific proteins are being modified to cause the changes in synaptic efficacy. We also know little about the molecular mechanisms underlying the structural changes in synapses that seem to accompany LTP and LTD. With the accumulation of evidence demonstrating that long-term synaptic modifications do in fact play important roles in a host of adaptive brain functions, we believe there is strong justification to continue efforts toward establishing the detailed molecular basis of the various forms of synaptic plasticity. This is particularly important because, as we have tried to make clear, such knowledge is likely to have a major impact on our understanding and treatment of a wide range of brain disorders.
What direction do we envision the field taking in the near future? By definition, synaptic plasticity is an electrophysiological phenomenon. Only by recording synaptic responses can the investigator be sure that synaptic function has been modified. Until relatively recently, most electrophysiological studies depended on pharmacological manipulations of proteins, an approach that was aided by the use of genetically modified mice. Thus far, gene knockout experiments addressing the function of ∼200 genes have been reported and close to 80% of these report effects on synaptic transmission or plasticity (Grant, 2006). With the growing consensus that NMDAR-dependent LTP is primarily mediated by an increase in postsynaptic AMPARs, while LTD is associated with loss of AMPARs from the PSD, there also has been an increased contribution of cell biologists who have focused on the molecular mechanisms underlying the changes in the levels of postsynaptic receptors as well as the changes in synaptic structure that occur. Indeed, a close interdependence has emerged between electrophysiologists and cell biologists and this has dramatically impacted the field. Thus, electrophysiologists are increasingly making use of tools that enable overexpression, knockdown, and molecular replacement of target proteins both in vitro and in vivo (eg, Schluter et al, 2006). These advances are enabling a true molecular dissection of the mechanisms underlying plasticity of mammalian brain synapses.
Recent advances in high-throughput technologies, such as gene-chip microarrays for mRNA profiling and advanced mass spectrometry for identification of multiple proteins in a mixed population, have enabled the identification of the molecular components of many biological phenomena and contributed significantly to understanding their regulation. Furthermore, the recent advent of ‘systems biology’ has contributed a number of conceptual advances that enable clearer interpretation of the complex interaction networks observed in signal transduction cascades (Alon, 2006). These advances have so far only lightly impacted the field of synaptic plasticity (Pocklington et al, 2006), but we envision that they will have increasing influence in the near future, despite the technical difficulties that are encountered when trying to apply these approaches to the inherently complex preparations used in the study of synaptic plasticity.
What might we gain from such studies? A clearer picture of the molecules which participate in the processes underlying the different forms of synaptic plasticity, and the dynamics of their activation and interactions, would enable the formulation of much more sophisticated models of the events underlying the triggering, expression, and maintenance of specific forms of synaptic plasticity. This in turn would create a strong foundation for focused efforts aimed at identifying the central pathways mediating synaptic plasticity and assaying their relevance to the phenomenon measured electrophysiologically.
By combining electrophysiological, molecular, cell biological and proteomic approaches, we envision that the next few decades will bring further clarification of the detailed molecular mechanisms underlying the major forms of synaptic plasticity that we have discussed in this review. The use of more sophisticated molecular and genetic manipulations, in particular, the development of mouse lines with highly restricted expression of transgenes that can be turned on or off with fine temporal control, will simultaneously facilitate the examination of the neural circuit basis of behavior. Together, we believe these approaches will allow sophisticated manipulation of synaptic plasticity mechanisms in highly restricted neural circuits and thereby greatly advance our understanding of how synaptic plasticity mediates both adaptive and pathological experience-dependent plasticity. Applying these same approaches to disease models should yield new insights into the molecular pathology of diseases of the synapse such as addiction, schizophrenia, and Alzheimer's diseases and also pave the way toward the development of novel and more efficacious treatments.
References
Abbott LF, Regehr WG (2004). Synaptic computation. Nature 431: 796–803.
Abraham WC, Bear MF (1996). Metaplasticity: the plasticity of synaptic plasticity. Trends Neurosci 19: 126–130. A review summarizing the mechanisms and functions of metaplasticity.
Abraham WC, Williams JM (2003). Properties and mechanisms of LTP maintenance. Neuroscientist 9: 463–474.
Ade KK, Lovinger DM (2007). Anandamide regulates postnatal development of long-term synaptic plasticity in the rat dorsolateral striatum. J Neurosci 27: 2403–2409.
Allen CB, Celikel T, Feldman DE (2003). Long-term depression induced by sensory deprivation during cortical map plasticity in vivo. Nat Neurosci 6: 291–299. Clear evidence that long-term depression is elicited in vivo during sensory deprivation.
Alon U (2006). An Introduction to Systems Biology: Design Principles of Biological Circuits. CRC Press.
Anwyl R (2006). Induction and expression mechanisms of postsynaptic NMDA receptor-independent homosynaptic long-term depression. Prog Neurobiol 78: 17–37.
Araque A, Carmignoto G, Haydon PG (2001). Dynamic signaling between astrocytes and neurons. Annu Rev Physiol 63: 795–813.
Armstrong JN, Saganich MJ, Xu NJ, Henkemeyer M, Heinemann SF, Contractor A (2006). B-ephrin reverse signaling is required for NMDA-independent long-term potentiation of mossy fibers in the hippocampus. J Neurosci 26: 3474–3481.
Ashby MC, De La Rue SA, Ralph GS, Uney J, Collingridge GL, Henley JM (2004). Removal of AMPA receptors (AMPARs) from synapses is preceded by transient endocytosis of extrasynaptic AMPARs. J Neurosci 24: 5172–5176.
Banke TG, Bowie D, Lee H, Huganir RL, Schousboe A, Traynelis SF (2000). Control of GluR1 AMPA receptor function by cAMP-dependent protein kinase. J Neurosci 20: 89–102.
Barria A, Muller D, Derkach V, Griffith LC, Soderling TR (1997). Regulatory phosphorylation of AMPA-type glutamate receptors by CaM-KII during long-term potentiation. Science 276: 2042–2045.
Bear MF, Cooper LN, Ebner FF (1987). A physiological basis for a theory of synapse modification. Science 237: 42–48.
Bear MF, Huber KM, Warren ST (2004). The mGluR theory of fragile X mental retardation. Trends Neurosci 27: 370–377.
Beattie EC, Carroll RC, Yu X, Morishita W, Yasuda H, von Zastrow M et al (2000). Regulation of AMPA receptor endocytosis by a signaling mechanism shared with LTD. Nat Neurosci 3: 1291–1300. Evidence that connects the signaling cascade involved in triggering LTD with endocytosis of AMPA receptors.
Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M et al (2002). Control of synaptic strength by glial TNFalpha. Science 295: 2282–2285.
Bellone C, Luscher C (2005). mGluRs induce a long-term depression in the ventral tegmental area that involves a switch of the subunit composition of AMPA receptors. Eur J Neurosci 21: 1280–1288.
Bellone C, Luscher C (2006). Cocaine triggered AMPA receptor redistribution is reversed in vivo by mGluR-dependent long-term depression. Nat Neurosci 9: 636–641.
Benke TA, Luthi A, Isaac JT, Collingridge GL (1998). Modulation of AMPA receptor unitary conductance by synaptic activity. Nature 393: 793–797. Evidence that LTP expression involves modification of the biophysical properties of AMPA receptors.
Betz WJ (1970). Depression of transmitter release at the neuromuscular junction of the frog. J Physiol 206: 629–644.
Bergles DE, Diamond JS, Jahr CE (1999). Clearance of glutamate inside the synapse and beyond. Curr Opin Neurobiol 9: 293–298.
Bienenstock EL, Cooper LN, Munro PW (1982). Theory for the development of neuron selectivity: orientation specificity and binocular interaction in visual cortex. J Neurosci 2: 32–48.
Blanpied TA, Scott DB, Ehlers MD (2002). Dynamics and regulation of clathrin coats at specialized endocytic zones of dendrites and spines. Neuron 36: 435–449. Localization of the sites of endocytosis in dendritic spines showing that they are adjacent to the PSD.
Bliss TV, Gardner-Medwin AR (1973). Long-lasting potentiation of synaptic transmission in the dentate area of the unanaestetized rabbit following stimulation of the perforant path. J Physiol 232: 357–374.
Bliss TV, Lomo T (1973). Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol 232: 331–356. Claassic papers demonstrating the existence of LTP in the hippocampus.
Blitzer RD, Connor JH, Brown GP, Wong T, Shenolikar S, Iyengar R et al (1998). Gating of CaMKII by cAMP-regulated protein phosphatase activity during LTP. Science 280: 1940–1942.
Boehm J, Kang MG, Johnson RC, Esteban J, Huganir RL, Malinow R (2006). Synaptic incorporation of AMPA receptors during LTP is controlled by a PKC phosphorylation site on GluR1. Neuron 51: 213–225.
Bolam JP, Hanley JJ, Booth PA, Bevan MD (2000). Synaptic organisation of the basal ganglia. J Anat 196: 527–542.
Bolshakov VY, Carboni L, Cobb MH, Siegelbaum SA, Belardetti F (2000). Dual MAP kinase pathways mediate opposing forms of long-term plasticity at CA3-CA1 synapses. Nat Neurosci 3: 1107–1112.
Bourne JN, Sorra KE, Hurlburt J, Harris KM (2007). Polyribosomes are increased in spines of CA1 dendrites 2 h after the induction of LTP in mature rat hippocampal slices. Hippocampus 17: 1–4.
Boyden ES, Katoh A, Raymond JL (2004). Cerebellum-dependent learning: the role of multiple plasticity mechanisms. Annu Rev Neurosci 27: 581–609.
Bramham CR, Messaoudi E (2005). BDNF function in adult synaptic plasticity: the synaptic consolidation hypothesis. Prog Neurobiol 76: 99–125.
Brebner K, Wong TP, Liu L, Liu Y, Campsall P, Gray S et al (2005). Nucleus accumbens long-term depression and the expression of behavioral sensitization. Science 310: 1340–1343.
Bredt DS, Nicoll RA (2003). AMPA receptor trafficking at excitatory synapses. Neuron 40: 361–379.
Brown TC, Tran IC, Backos DS, Esteban JA (2005). NMDA receptor-dependent activation of the small GTPase Rab5 drives the removal of synaptic AMPA receptors during hippocampal LTD. Neuron 45: 81–94.
Burrone J, Murthy VN (2003). Synaptic gain control and homeostasis. Curr Opin Neurobiol 13: 560–567.
Burrone J, O’Byrne M, Murthy VN (2002). Multiple forms of synaptic plasticity triggered by selective suppression of activity in individual neurons. Nature 420: 414–418.
Carroll RC, Beattie EC, von Zastrow M, Malenka RC (2001). Role of AMPA receptor endocytosis in synaptic plasticity. Nat Rev Neurosci 2: 315–324.
Carroll RC, Beattie EC, Xia H, Luscher C, Altschuler Y, Nicoll RA et al (1999). Dynamin-dependent endocytosis of ionotropic glutamate receptors. Proc Natl Acad Sci USA 96: 14112–14117.
Castillo PE, Janz R, Sudhof TC, Tzounopoulos T, Malenka RC, Nicoll RA (1997). Rab3A is essential for mossy fibre long-term potentiation in the hippocampus. Nature 388: 590–593.
Castillo PE, Schoch S, Schmitz F, Sudhof TC, Malenka RC (2002). RIM1alpha is required for presynaptic long-term potentiation. Nature 415: 327–330. Papers demonstrating the importance of specific proteins for presynaptic LTP.
Castro-Alamancos MA, Calcagnotto ME (1999). Presynaptic long-term potentiation in corticothalamic synapses. J Neurosci 19: 9090–9097.
Celikel T, Szostak VA, Feldman DE (2004). Modulation of spike timing by sensory deprivation during induction of cortical map plasticity. Nat Neurosci 7: 534–541.
Chen L, Chetkovich DM, Petralia RS, Sweeney NT, Kawasaki Y, Wenthold RJ et al (2000). Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature 408: 936–943. Experiments demonstrating that the TARP stargazin is required for the trafficking of AMPARs to synapses.
Chevaleyre V, Castillo PE (2004). Endocannabinoid-mediated metaplasticity in the hippocampus. Neuron 43: 871–881.
Chevaleyre V, Takahashi KA, Castillo PE (2006). Endocannabinoid-mediated synaptic plasticity in the CNS. Annu Rev Neurosci 29: 37–76.
Choi S, Lovinger DM (1997). Decreased probability of neurotransmitter release underlies striatal long-term depression and postnatal development of corticostriatal synapses. Proc Natl Acad Sci USA 94: 2665–2670.
Chung HJ, Steinberg JP, Huganir RL, Linden DJ (2003). Requirement of AMPA receptor GluR2 phosphorylation for cerebellar long-term depression. Science 300: 1751–1755. Demonstration of the importance of specific GluR2 phosphorylation sites for cerebellar LTD.
Colledge M, Snyder EM, Crozier RA, Soderling JA, Jin Y, Langeberg LK et al (2003). Ubiquitination regulates PSD-95 degradation and AMPA receptor surface expression. Neuron 40: 595–607.
Collingridge GL, Isaac JT, Wang YT (2004). Receptor trafficking and synaptic plasticity. Nat Rev Neurosci 5: 952–962.
Contractor A, Rogers C, Maron C, Henkemeyer M, Swanson GT, Heinemann SF (2002). Trans-synaptic Eph receptor-ephrin signaling in hippocampal mossy fiber LTP. Science 296: 1864–1869.
Cottrell JR, Borok E, Horvath TL, Nedivi E (2004). CPG2: a brain- and synapse-specific protein that regulates the endocytosis of glutamate receptors. Neuron 44: 677–690.
Coyle JT, Tsai G (2004). NMDA receptor function, neuroplasticity, and the pathophysiology of schizophrenia. Int Rev Neurobiol 59: 491–515.
Crozier RA, Wang Y, Liu CH, Bear MF (2007). Deprivation-induced synaptic depression by distinct mechanisms in different layers of mouse visual cortex. Proc Natl Acad Sci USA 104: 1383–1388.
Cummings JA, Mulkey RM, Nicoll RA, Malenka RC (1996). Ca2+ signaling requirements for long-term depression in the hippocampus. Neuron 16: 825–833.
Dan Y, Poo MM (2006). Spike timing-dependent plasticity: from synapse to perception. Physiol Rev 86: 1033–1048.
Danbolt NC (2001). Glutamate uptake. Prog Neurobiol 65: 1–105.
De Camilli P, Benfenati F, Valtorta F, Greengard P (1990). The synapsins. Annu Rev Cell Biol 6: 433–460.
De Zeeuw CI, Hansel C, Bian F, Koekkoek SK, van Alphen AM, Linden DJ et al (1998). Expression of a protein kinase C inhibitor in Purkinje cells blocks cerebellar LTD and adaptation of the vestibulo-ocular reflex. Neuron 20: 495–508.
Derkach V, Barria A, Soderling TR (1999). Ca2+/calmodulin-kinase II enhances channel conductance of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate type glutamate receptors. Proc Natl Acad Sci USA 96: 3269–3274.
Derkach VA, Oh MC, Guire ES, Soderling TR (2007). Regulatory mechanisms of AMPA receptors in synaptic plasticity. Nat Rev Neurosci 8: 101–113.
Dobrunz LE, Stevens CF (1997). Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron 18: 995–1008.
Dong Y, Saal D, Thomas M, Faust R, Bonci A, Robinson T et al (2004). Cocaine-induced potentiation of synaptic strength in dopamine neurons: behavioral correlates in GluRA(−/−) mice. Proc Natl Acad Sci USA 101: 14282–14287.
Drake CT, Terman GW, Simmons ML, Milner TA, Kunkel DD, Schwartzkroin PA et al (1994). Dynorphin opioids present in dentate granule cells may function as retrograde inhibitory neurotransmitters. J Neurosci 14: 3736–3750.
Du J, Gray NA, Falke C, Yuan P, Szabo S, Manji HK (2003). Structurally dissimilar antimanic agents modulate synaptic plasticity by regulating AMPA glutamate receptor subunit GluR1 synaptic expression. Ann N Y Acad Sci 1003: 378–380.
Du J, Gray NA, Falke CA, Chen W, Yuan P, Szabo ST et al (2004). Modulation of synaptic plasticity by antimanic agents: the role of AMPA glutamate receptor subunit 1 synaptic expression. J Neurosci 24: 6578–6589.
Du J, Suzuki K, Wei Y, Wang Y, Blumenthal R, Chen Z et al (2007). The anticonvulsants lamotrigine, riluzole, and valproate differentially regulate AMPA receptor membrane localization: relationship to clinical effects in mood disorders. Neuropsychopharmacology 32: 793–802.
Dudek SM, Bear MF (1992). Homosynaptic long-term depression in area CA1 of hippocampus and effects of N-methyl-D-aspartate receptor blockade. Proc Natl Acad Sci USA 89: 4363–4367. First demonstration of NMDAR-dependent LTD in the hippocampus.
Durand GM, Kovalchuk Y, Konnerth A (1996). Long-term potentiation and functional synapse induction in developing hippocampus. Nature 381: 71–75.
Ehlers MD (2000). Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron 28: 511–525.
Ehrlich I, Klein M, Rumpel S, Malinow R (2007). PSD-95 is required for activity-driven synapse stabilization. Proc Natl Acad Sci USA 104: 4176–4181.
Ehrlich I, Malinow R (2004). Postsynaptic density 95 controls AMPA receptor incorporation during long-term potentiation and experience-driven synaptic plasticity. J Neurosci 24: 916–927.
El-Husseini AE, Schnell E, Dakoji S, Sweeney N, Zhou Q, Prange O et al (2002). Synaptic strength regulated by palmitate cycling on PSD-95. Cell 108: 849–863.
Elias GM, Funke L, Stein V, Grant SG, Bredt DS, Nicoll RA (2006). Synapse-specific and developmentally regulated targeting of AMPA receptors by a family of MAGUK scaffolding proteins. Neuron 52: 307–320.
Esteban JA, Shi SH, Wilson C, Nuriya M, Huganir RL, Malinow R (2003). PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat Neurosci 6: 136–143.
Everitt BJ, Robbins TW (2005). Neural systems of reinforcement for drug addiction: from actions to habits to compulsion. Nat Neurosci 8: 1481–1489.
Everitt BJ, Wolf ME (2002). Psychomotor stimulant addiction: a neural systems perspective. J Neurosci 22: 3312–3320.
Faleiro LJ, Jones S, Kauer JA (2004). Rapid synaptic plasticity of glutamatergic synapses on dopamine neurons in the ventral tegmental area in response to acute amphetamine injection. Neuropsychopharmacology 29: 2115–2125.
Feinmark SJ, Begum R, Tsvetkov E, Goussakov I, Funk CD, Siegelbaum SA et al (2003). 12-Lipoxygenase metabolites of arachidonic acid mediate metabotropic glutamate receptor-dependent long-term depression at hippocampal CA3-CA1 synapses. J Neurosci 23: 11427–11435.
Foeller E, Feldman DE (2004). Synaptic basis for developmental plasticity in somatosensory cortex. Curr Opin Neurobiol 14: 89–95.
Fourgeaud L, Mato S, Bouchet D, Hemar A, Worley PF, Manzoni OJ (2004). A single in vivo exposure to cocaine abolishes endocannabinoid-mediated long-term depression in the nucleus accumbens. J Neurosci 24: 6939–6945.
Frenkel MY, Bear MF (2004). How monocular deprivation shifts ocular dominance in visual cortex of young mice. Neuron 44: 917–923.
Freund TF, Katona I, Piomelli D (2003). Role of endogenous cannabinoids in synaptic signaling. Physiol Rev 83: 1017–1066.
Frey U, Morris RG (1997). Synaptic tagging and long-term potentiation. Nature 385: 533–536. Presentation of the synaptic tagging hypothesis for the maintenance of LTP.
Fukunaga K, Muller D, Miyamoto E (1995). Increased phosphorylation of Ca2+/calmodulin-dependent protein kinase II and its endogenous substrates in the induction of long-term potentiation. J Biol Chem 270: 6119–6124.
Futai K, Kim MJ, Hashikawa T, Scheiffele P, Sheng M, Hayashi Y (2007). Retrograde modulation of presynaptic release probability through signaling mediated by PSD-95-neuroligin. Nat Neurosci 10: 186–195.
Gallagher SM, Daly CA, Bear MF, Huber KM (2004). Extracellular signal-regulated protein kinase activation is required for metabotropic glutamate receptor-dependent long-term depression in hippocampal area CA1. J Neurosci 24: 4859–4864.
Gerber DJ, Hall D, Miyakawa T, Demars S, Gogos JA, Karayiorgou M et al (2003). Evidence for association of schizophrenia with genetic variation in the 8p21.3 gene, PPP3CC, encoding the calcineurin gamma subunit. ProcNatl Acad Sci USA 100: 8993–8998.
Gerdeman GL, Partridge JG, Lupica CR, Lovinger DM (2003). It could be habit forming: drugs of abuse and striatal synaptic plasticity. Trends Neurosci 26: 184–192.
Gerdeman GL, Ronesi J, Lovinger DM (2002). Postsynaptic endocannabinoid release is critical to long-term depression in the striatum. Nat Neurosci 5: 446–451. Demonstration that LTD in the striatum requires postsynaptic release of endocannabinoids.
Geschwind DH, Levitt P (2007). Autism spectrum disorders: developmental disconnection syndromes. Curr Opin Neurobiol 17: 103–111.
Giese KP, Fedorov NB, Filipkowski RK, Silva AJ (1998). Autophosphorylation at Thr286 of the alpha calcium-calmodulin kinase II in LTP and learning. Science 279: 870–873. Elegant genetic manipulation of CaMKII demonstrating its importance in LTP and learning.
Gomez LL, Alam S, Smith KE, Horne E, Dell’Acqua ML (2002). Regulation of A-kinase anchoring protein 79/150-cAMP-dependent protein kinase postsynaptic targeting by NMDA receptor activation of calcineurin and remodeling of dendritic actin. J Neurosci 22: 7027–7044.
Grant SG (2006). The synapse proteome and phosphoproteome: a new paradigm for synapse biology. Biochem Soc Trans 34: 59–63.
Gray NA, Du J, Falke CS, Yuan P, Manji HK (2003). Lithium regulates total and synaptic expression of the AMPA glutamate receptor GluR2 in vitro and in vivo. Ann NY Acad Sci 1003: 402–404.
Groc L, Heine M, Cognet L, Brickley K, Stephenson FA, Lounis B et al (2004). Differential activity-dependent regulation of the lateral mobilities of AMPA and NMDA receptors. Nat Neurosci 7: 695–696.
Grooms SY, Noh KM, Regis R, Bassell GJ, Bryan MK, Carroll RC et al (2006). Activity bidirectionally regulates AMPA receptor mRNA abundance in dendrites of hippocampal neurons. J Neurosci 26: 8339–8351.
Gubellini P, Pisani A, Centonze D, Bernardi G, Calabresi P (2004). Metabotropic glutamate receptors and striatal synaptic plasticity: implications for neurological diseases. Prog Neurobiol 74: 271–300.
Hanley JG, Henley JM (2005). PICK1 is a calcium-sensor for NMDA-induced AMPA receptor trafficking. EMBO J 24: 3266–3278.
Harney SC, Rowan M, Anwyl R (2006). Long-term depression of NMDA receptor-mediated synaptic transmission is dependent on activation of metabotropic glutamate receptors and is altered to long-term potentiation by low intracellular calcium buffering. J Neurosci 26: 1128–1132.
Harris GC, Aston-Jones G (2003). Critical role for ventral tegmental glutamate in preference for a cocaine-conditioned environment. Neuropsychopharmacology 28: 73–76.
Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R (2000). Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science 287: 2262–2267. Clever experiments showing that LTP involves the synaptic insertion of GluR1-containing AMPA receptors.
Haydon PG (2001). GLIA: listening and talking to the synapse. Nat Rev Neurosci 2: 185–193.
Hebb DO (1949). The Organization of Behavior. John Wiley: New York. Classic text pointing out the theoretical importance of ‘LTP’ -like synaptic modifications.
Heynen AJ, Quinlan EM, Bae DC, Bear MF (2000). Bidirectional, activity-dependent regulation of glutamate receptors in the adult hippocampus in vivo. Neuron 28: 527–536.
Heynen AJ, Yoon BJ, Liu CH, Chung HJ, Huganir RL, Bear MF (2003). Molecular mechanism for loss of visual cortical responsiveness following brief monocular deprivation. Nat Neurosci 6: 854–862.
Hofmann SG, Meuret AE, Smits JA, Simon NM, Pollack MH, Eisenmenger K et al (2006). Augmentation of exposure therapy with D-cycloserine for social anxiety disorder. Arch Gene Psychiatry 63: 298–304.
Hou L, Antion MD, Hu D, Spencer CM, Paylor R, Klann E (2006). Dynamic translational and proteasomal regulation of fragile X mental retardation protein controls mGluR-dependent long-term depression. Neuron 51: 441–454.
Hou L, Klann E (2004). Activation of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway is required for metabotropic glutamate receptor-dependent long-term depression. J Neurosci 24: 6352–6361.
Hrabetova S, Sacktor TC (1996). Bidirectional regulation of protein kinase M zeta in the maintenance of long-term potentiation and long-term depression. J Neurosci 16: 5324–5333.
Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S et al (2006). AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron 52: 831–843.
Huang CC, Hsu KS (2006). Sustained activation of metabotropic glutamate receptor 5 and protein tyrosine phosphatases mediate the expression of (S)-3,5-dihydroxyphenylglycine-induced long-term depression in the hippocampal CA1 region. J Neurochem 96: 179–194.
Huang YY, Colino A, Selig DK, Malenka RC (1992). The influence of prior synaptic activity on the induction of long-term potentiation. Science 255: 730–733.
Huber KM, Gallagher SM, Warren ST, Bear MF (2002). Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci USA 99: 7746–7750.
Huber KM, Kayser MS, Bear MF (2000). Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science 288: 1254–1257. Papers demonstrating that mGluR LTD requires protein synthesis and may play a role in Fragile X syndrome.
Huber KM, Roder JC, Bear MF (2001). Chemical induction of mGluR5- and protein synthesis-dependent long-term depression in hippocampal area CA1. J Neurophysiol 86: 321–325.
Hyman SE, Malenka RC (2001). Addiction and the brain: the neurobiology of compulsion and its persistence. Nat Rev Neurosci 2: 695–703.
Hyman SE, Malenka RC, Nestler EJ (2006). Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci 29: 565–598.
Isaac JT, Nicoll RA, Malenka RC (1995). Evidence for silent synapses: implications for the expression of LTP. Neuron 15: 427–434. Direct demonstration of the existence of silent synapses and their conversion to functional synapses during LTP.
Ito M (1989). Long-term depression. Annu Rev Neurosci 12: 85–102.
Ito M (2002). The molecular organization of cerebellar long-term depression. Nat Rev Neurosci 3: 896–902.
Javitt DC (2006). Is the glycine site half saturated or half unsaturated? Effects of glutamatergic drugs in schizophrenia patients. Curr Opin Psychiatry 19: 151–157.
Jia Z, Agopyan N, Miu P, Xiong Z, Henderson J, Gerlai R et al (1996). Enhanced LTP in mice deficient in the AMPA receptor GluR2. Neuron 17: 945–956.
Job C, Eberwine J (2001). Localization and translation of mRNA in dendrites and axons. Nat Rev Neurosci 2: 889–898.
Jorntell H, Hansel C (2006). Synaptic memories upside down: bidirectional plasticity at cerebellar parallel fiber-Purkinje cell synapses. Neuron 52: 227–238.
Ju W, Morishita W, Tsui J, Gaietta G, Deerinck TJ, Adams SR et al (2004). Activity-dependent regulation of dendritic synthesis and trafficking of AMPA receptors. Nat Neurosci 7: 244–253.
Kalia LV, Gingrich JR, Salter MW (2004). Src in synaptic transmission and plasticity. Oncogene 23: 8007–8016.
Kameyama K, Lee HK, Bear MF, Huganir RL (1998). Involvement of a postsynaptic protein kinase A substrate in the expression of homosynaptic long-term depression. Neuron 21: 1163–1175.
Kamiya H, Umeda K, Ozawa S, Manabe T (2002). Presynaptic Ca2+ entry is unchanged during hippocampal mossy fiber long-term potentiation. J Neurosci 22: 10524–10528.
Kandel ER, Tauc L (1965). Mechanism of heterosynaptic facilitation in the giant cell of the abdominal ganglion of Aplysia depilans. J Physiol 181: 28–47.
Karmarkar UR, Dan Y (2006). Experience-dependent plasticity in adult visual cortex. Neuron 52: 577–585.
Kastning K, Kukhtina V, Kittler JT, Chen G, Pechstein A, Enders S et al (2007). Molecular determinants for the interaction between AMPA receptors and the clathrin adaptor complex AP-2. Proc Natl Acad Sci USA 104: 2991–2996.
Katz B, Miledi R (1968). The role of calcium in neuromuscular facilitation. J Physiol 195: 481–492.
Kauer JA (2004). Learning mechanisms in addiction: synaptic plasticity in the ventral tegmental area as a result of exposure to drugs of abuse. Annu Rev Physiol 66: 447–475.
Kelley AE (2004). Memory and addiction: shared neural circuitry and molecular mechanisms. Neuron 44: 161–179.
Kelz MB, Chen J, Carlezon Jr WA, Whisler K, Gilden L, Beckmann AM et al (1999). Expression of the transcription factor deltaFosB in the brain controls sensitivity to cocaine. Nature 401: 272–276.
Kim CH, Chung HJ, Lee HK, Huganir RL (2001). Interaction of the AMPA receptor subunit GluR2/3 with PDZ domains regulates hippocampal long-term depression. Proc Natl Acad Sci USA 98: 11725–11730.
Kim E, Sheng M (2004). PDZ domain proteins of synapses. Nat Rev Neurosci 5: 771–781.
Kirkwood A, Bear MF (1994). Homosynaptic long-term depression in the visual cortex. J Neurosci 14: 3404–3412.
Kirkwood A, Dudek SM, Gold JT, Aizenman CD, Bear MF (1993). Common forms of synaptic plasticity in the hippocampus and neocortex in vitro. Science 260: 1518–1521.
Koekkoek SK, Yamaguchi K, Milojkovic BA, Dortland BR, Ruigrok TJ, Maex R et al (2005). Deletion of FMR1 in Purkinje cells enhances parallel fiber LTD, enlarges spines, and attenuates cerebellar eyelid conditioning in Fragile X syndrome. Neuron 47: 339–352.
Kombian SB, Mouginot D, Pittman QJ (1997). Dendritically released peptides act as retrograde modulators of afferent excitation in the supraoptic nucleus in vitro. Neuron 19: 903–912.
Kornau HC, Schenker LT, Kennedy MB, Seeburg PH (1995). Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science 269: 1737–1740. Identification of PSD-95 as a major synaptic scaffolding protein.
Kreitzer AC, Malenka RC (2005). Dopamine modulation of state-dependent endocannabinoid release and long-term depression in the striatum. J Neurosci 25: 10537–10545.
Kreitzer AC, Malenka RC (2007). Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson's disease models. Nature 445: 643–647. Demonstration that endocannabinoid-mediated LTD may play an important role in Parkinson's disease.
Kreitzer AC, Regehr WG (2002). Retrograde signaling by endocannabinoids. Curr Opin Neurobiol 12: 324–330.
Lai MM, Hong JJ, Ruggiero AM, Burnett PE, Slepnev VI, De Camilli P et al (1999). The calcineurin–dynamin 1 complex as a calcium sensor for synaptic vesicle endocytosis. J Biol Chem 274: 25963–25966.
Lauri SE, Bortolotto ZA, Bleakman D, Ornstein PL, Lodge D, Isaac JT et al (2001). A critical role of a facilitatory presynaptic kainate receptor in mossy fiber LTP. Neuron 32: 697–709.
Lee HK, Barbarosie M, Kameyama K, Bear MF, Huganir RL (2000). Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature 405: 955–959. Demonstration of potential importance of AMPA receptor phosphorylation and dephosphorylation for LTP and LTD.
Lee HK, Kameyama K, Huganir RL, Bear MF (1998). NMDA induces long-term synaptic depression and dephosphorylation of the GluR1 subunit of AMPA receptors in hippocampus. Neuron 21: 1151–1162.
Lee HK, Takamiya K, Han JS, Man H, Kim CH, Rumbaugh G et al (2003). Phosphorylation of the AMPA receptor GluR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell 112: 631–643.
Lee SH, Liu L, Wang YT, Sheng M (2002). Clathrin adaptor AP2 and NSF interact with overlapping sites of GluR2 and play distinct roles in AMPA receptor trafficking and hippocampal LTD. Neuron 36: 661–674.
Lewis DA, Levitt P (2002). Schizophrenia as a disorder of neurodevelopment. Annu Rev Neurosci 25: 409–432.
Li XM, Li CC, Yu SS, Chen JT, Sabapathy K, Ruan DY (2007). JNK1 contributes to metabotropic glutamate receptor-dependent long-term depression and short-term synaptic plasticity in the mice area hippocampal CA1. Eur J Neurosci 25: 391–396.
Liao D, Hessler NA, Malinow R (1995). Activation of postsynaptically silent synapses during pairing-induced LTP in CA1 region of hippocampal slice. Nature 375: 400–404. Direct demonstration of the existence of silent synapses and their conversion to functional synapses during LTP.
Lin JW, Ju W, Foster K, Lee SH, Ahmadian G, Wyszynski M et al (2000). Distinct molecular mechanisms and divergent endocytotic pathways of AMPA receptor internalization. Nat Neurosci 3: 1282–1290.
Linden DJ (1997). Long-term potentiation of glial synaptic currents in cerebellar culture. Neuron 18: 983–994.
Linden DJ, Connor JA (1991). Participation of postsynaptic PKC in cerebellar long-term depression in culture. Science 254: 1656–1659.
Linden DJ, Connor JA (1995). Long-term synaptic depression. Annu Rev Neurosci 18: 319–357.
Ling DS, Benardo LS, Serrano PA, Blace N, Kelly MT, Crary JF et al (2002). Protein kinase Mzeta is necessary and sufficient for LTP maintenance. Nat Neurosci 5: 295–296.
Lisman J (1989). A mechanism for the Hebb and the anti-Hebb processes underlying learning and memory. Proc Natl Acad Sci USA 86: 9574–9578. Presentation of influential hypothesis of the signal transduction cascades triggering LTP and LTD.
Lisman JE, Harris KM (1993). Quantal analysis and synaptic anatomy—integrating two views of hippocampal plasticity. Trends Neurosci 16: 141–147.
Lisman JE, Zhabotinsky AM (2001). A model of synaptic memory: a CaMKII/PP1 switch that potentiates transmission by organizing an AMPA receptor anchoring assembly. Neuron 31: 191–201.
Lissin DV, Gomperts SN, Carroll RC, Christine CW, Kalman D, Kitamura M et al (1998). Activity differentially regulates the surface expression of synaptic AMPA and NMDA glutamate receptors. Proc Natl Acad Sci USA 95: 7097–7102. First demonstration that activity can independently influence the expression of synaptic AMPA receptors and NMDA receptors.
Llano I, Leresche N, Marty A (1991). Calcium entry increases the sensitivity of cerebellar Purkinje cells to applied GABA and decreases inhibitory synaptic currents. Neuron 6: 565–574.
Lledo PM, Hjelmstad GO, Mukherji S, Soderling TR, Malenka RC, Nicoll RA (1995). Calcium/calmodulin-dependent kinase II and long-term potentiation enhance synaptic transmission by the same mechanism. Proc Natl Acad Sci USA 92: 11175–11179.
Lonart G, Janz R, Johnson KM, Sudhof TC (1998). Mechanism of action of rab3A in mossy fiber LTP. Neuron 21: 1141–1150.
Lonart G, Schoch S, Kaeser PS, Larkin CJ, Sudhof TC, Linden DJ (2003). Phosphorylation of RIM1alpha by PKA triggers presynaptic long-term potentiation at cerebellar parallel fiber synapses. Cell 115: 49–60.
Luscher C, Nicoll RA, Malenka RC, Muller D (2000). Synaptic plasticity and dynamic modulation of the postsynaptic membrane. Nat Neurosci 3: 545–550.
Luscher C, Xia H, Beattie EC, Carroll RC, von Zastrow M, Malenka RC et al (1999). Role of AMPA receptor cycling in synaptic transmission and plasticity. Neuron 24: 649–658.
Lynch MA (2004). Long-term potentiation and memory. Physiol Rev 84: 87–136.
MacDermott AB, Role LW, Siegelbaum SA (1999). Presynaptic ionotropic receptors and the control of transmitter release. Annu Rev Neurosci 22: 443–485.
Magleby KL, Zengel JE (1982). A quantitative description of stimulation-induced changes in transmitter release at the frog neuromuscular junction. J Gen Physiol 80: 613–638.
Makhinson M, Chotiner JK, Watson JB, O’Dell TJ (1999). Adenylyl cyclase activation modulates activity-dependent changes in synaptic strength and Ca2+/calmodulin-dependent kinase II autophosphorylation. J Neurosci 19: 2500–2510.
Malenka RC (1991). Postsynaptic factors control the duration of synaptic enhancement in area CA1 of the hippocampus. Neuron 6: 53–60.
Malenka RC, Bear MF (2004). LTP and LTD: an embarrassment of riches. Neuron 44: 5–21.
Malenka RC, Kauer JA, Perkel DJ, Mauk MD, Kelly PT, Nicoll RA et al (1989). An essential role for postsynaptic calmodulin and protein kinase activity in long-term potentiation. Nature 340: 554–557.
Malenka RC, Nicoll RA (1993). NMDA-receptor-dependent synaptic plasticity: multiple forms and mechanisms. Trends in Neurosci 16: 521–527.
Malenka RC, Nicoll RA (1997). Silent synapses speak up. Neuron 19: 473–476.
Malenka RC, Nicoll RA (1999). Long-term potentiation—a decade of progress? Science 285: 1870–1874.
Malinow R, Malenka RC (2002). AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci 25: 103–126.
Malinow R, Schulman H, Tsien RW (1989). Inhibition of postsynaptic PKC or CaMKII blocks induction but not expression of LTP. Science 245: 862–866.
Markram H, Lubke J, Frotscher M, Sakmann B (1997). Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science 275: 213–215.
Martin M, Chen BT, Hopf FW, Bowers MS, Bonci A (2006). Cocaine self-administration selectively abolishes LTD in the core of the nucleus accumbens. Nat Neurosci 9: 868–869.
Martin SJ, Grimwood PD, Morris RG (2000). Synaptic plasticity and memory: an evaluation of the hypothesis. Annu Rev Neurosci 23: 649–711. A review of the evidence that LTP plays a role in learning and memory.
Mato S, Chevaleyre V, Robbe D, Pazos A, Castillo PE, Manzoni OJ (2004). A single in-vivo exposure to delta 9THC blocks endocannabinoid-mediated synaptic plasticity. Nat Neurosci 7: 585–586.
Matsuzaki M, Ellis-Davies GC, Nemoto T, Miyashita Y, Iino M, Kasai H (2001). Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat Neurosci 4: 1086–1092.
Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H (2004). Structural basis of long-term potentiation in single dendritic spines. Nature 429: 761–766. Demonstration of morphological changes in dendritic spines during LTP.
Mayer ML, Westbrook GL, Guthrie PB (1984). Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature 309: 261–263. Classic paper describing the unusual biophysical properties of NMDA receptors.
Mayford M, Baranes D, Podsypanina K, Kandel ER (1996). The 3′-untranslated region of CaMKII alpha is a cis-acting signal for the localization and translation of mRNA in dendrites. Proc Natl Acad Sci USA 93: 13250–13255.
McCullumsmith RE, Clinton SM, Meador-Woodruff JH (2004). Schizophrenia as a disorder of neuroplasticity. Int Rev Neurobiol 59: 19–45.
McKernan MG, Shinnick-Gallagher P (1997). Fear conditioning induces a lasting potentiation of synaptic currents in vitro. Nature 390: 607–611. Paper demonstrating that fear conditioning elicits LTP in the amygdala.
Meng Y, Zhang Y, Jia Z (2003). Synaptic transmission and plasticity in the absence of AMPA glutamate receptor GluR2 and GluR3. Neuron 39: 163–176.
Miller RJ (1998). Presynaptic receptors. Annu Rev Pharmacol Toxicol 38: 201–227.
Montgomery JM, Zamorano PL, Garner CC (2004). MAGUKs in synapse assembly and function: an emerging view. Cell Mol Life Sci 61: 911–929.
Morishita W, Connor JH, Xia H, Quinlan EM, Shenolikar S, Malenka RC (2001). Regulation of synaptic strength by protein phosphatase 1. Neuron 32: 1133–1148.
Morishita W, Marie H, Malenka RC (2005). Distinct triggering and expression mechanisms underlie LTD of AMPA and NMDA synaptic responses. Nat Neurosci 8: 1043–1050.
Morris RG (2006). Elements of a neurobiological theory of hippocampal function: the role of synaptic plasticity, synaptic tagging and schemas. Eur J Neurosci 23: 2829–2846.
Morris RG, Frey U (1997). Hippocampal synaptic plasticity: role in spatial learning or the automatic recording of attended experience? Philos Trans R Soc Lond 352: 1489–1503.
Moult PR, Gladding CM, Sanderson TM, Fitzjohn SM, Bashir ZI, Molnar E et al (2006). Tyrosine phosphatases regulate AMPA receptor trafficking during metabotropic glutamate receptor-mediated long-term depression. J Neurosci 26: 2544–2554.
Mulkey RM, Endo S, Shenolikar S, Malenka RC (1994). Involvement of a calcineurin/inhibitor-1 phosphatase cascade in hippocampal long-term depression. Nature 369: 486–488.
Mulkey RM, Herron CE, Malenka RC (1993). An essential role for protein phosphatases in hippocampal long-term depression. Science 261: 1051–1055.
Mulkey RM, Malenka RC (1992). Mechanisms underlying induction of homosynaptic long-term depression in area CA1 of the hippocampus. Neuron 9: 967–975.
Myers KM, Davis M (2002). Behavioral and neural analysis of extinction. Neuron 36: 567–584.
Nagerl UV, Eberhorn N, Cambridge SB, Bonhoeffer T (2004). Bidirectional activity-dependent morphological plasticity in hippocampal neurons. Neuron 44: 759–767.
Naggapan G, Lu B (2005). Activity-dependent modulation of the BDNF receptor TrkB: mechanisms and implications. Trends Neurosci 28: 464–471.
Nicoll RA (2003). Expression mechanisms underlying long-term potentiation: a postsynaptic view. Philos Trans R Soc Lond 358: 721–726.
Nicoll RA, Kauer JA, Malenka RC (1988). The current excitement in long-term potentiation. Neuron 1: 97–103.
Nicoll RA, Malenka RC (1995). Contrasting properties of two forms of long-term potentiation in the hippocampus. Nature 377: 115–118.
Nicoll RA, Schmitz D (2005). Synaptic plasticity at hippocampal mossy fibre synapses. Nat Rev Neurosci 6: 863–876.
Nicoll RA, Tomita S, Bredt DS (2006). Auxiliary subunits assist AMPA-type glutamate receptors. Science 311: 1253–1256.
Nishiyama M, Hong K, Mikoshiba K, Poo MM, Kato K (2000). Calcium stores regulate the polarity and input specificity of synaptic modification. Nature 408: 584–588.
Nosyreva ED, Huber KM (2005). Developmental switch in synaptic mechanisms of hippocampal metabotropic glutamate receptor-dependent long-term depression. J Neurosci 25: 2992–3001.
Nosyreva ED, Huber KM (2006). Metabotropic receptor-dependent long-term depression persists in the absence of protein synthesis in the mouse model of fragile X syndrome. J Neurophysiol 95: 3291–3295.
Nowak L, Bregestovski P, Ascher P, Herbet A, Prochiantz A (1984). Magnesium gates glutamate-activated channels in mouse central neurones. Nature 307: 462–465. Classic paper describing the unusual biophysical properties of NMDA receptors.
Nugent FS, Penick EC, Kauer JA (2007). Opioids block long-term potentiation of inhibitory synapses. Nature 446: 1086–1090.
Nusser Z, Lujan R, Laube G, Roberts JD, Molnar E, Somogyi P (1998). Cell type and pathway dependence of synaptic AMPA receptor number and variability in the hippocampus. Neuron 21: 545–559.
Oh MC, Derkach VA (2005). Dominant role of the GluR2 subunit in regulation of AMPA receptors by CaMKII. Nat Neurosci 8: 853–854.
Oliet SH, Malenka RC, Nicoll RA (1997). Two distinct forms of long-term depression coexist in CA1 hippocampal pyramidal cells. Neuron 18: 969–982.
Otto MW, Basden SL, Leyro TM, McHugh RK, Hofmann SG (2007). Clinical perspectives on the combination of D-cycloserine and cognitive-behavioral therapy for the treatment of anxiety disorders. CNS Spectr 12: 51–56 59–61.
Palmer CL, Lim W, Hastie PG, Toward M, Korolchuk VI, Burbidge SA et al (2005). Hippocalcin functions as a calcium sensor in hippocampal LTD. Neuron 47: 487–494.
Park M, Penick EC, Edwards JG, Kauer JA, Ehlers MD (2004). Recycling endosomes supply AMPA receptors for LTP. Science 305: 1972–1975. Experiments defining the intracellular compartment that supplies AMPARs to synapses during LTP.
Park M, Salgado JM, Ostroff L, Helton TD, Robinson CG, Harris KM et al (2006). Plasticity-induced growth of dendritic spines by exocytic trafficking from recycling endosomes. Neuron 52: 817–830.
Passafaro M, Piech V, Sheng M (2001). Subunit-specific temporal and spatial patterns of AMPA receptor exocytosis in hippocampal neurons. Nat Neurosci 4: 917–926.
Pastalkova E, Serrano P, Pinkhasova D, Wallace E, Fenton AA, Sacktor TC (2006). Storage of spatial information by the maintenance mechanism of LTP. Science 313: 1141–1144. Evidence that LTP maintenance is required for an engram in the hippocampus.
Pavlov IP (1927). Conditioned Reflexes: An Investigation of the Physiological Activity of the Cerebral Cortex. Oxford University Press: London.
Peineau S, Taghibiglou C, Bradley C, Wong TP, Liu L, Lu J et al (2007). LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron 53: 703–717.
Perez JL, Khatri L, Chang C, Srivastava S, Osten P, Ziff EB (2001). PICK1 targets activated protein kinase Calpha to AMPA receptor clusters in spines of hippocampal neurons and reduces surface levels of the AMPA-type glutamate receptor subunit 2. J Neurosci 21: 5417–5428.
Perez-Otano I, Ehlers MD (2005). Homeostatic plasticity and NMDA receptor trafficking. Trends Neurosci 28: 229–238.
Pettit DL, Perlman S, Malinow R (1994). Potentiated transmission and prevention of further LTP by increased CaMKII activity in postsynaptic hippocampal slice neurons. Science 266: 1881–1885.
Pfeiffer BE, Huber KM (2006). Current advances in local protein synthesis and synaptic plasticity. J Neurosci 26: 7147–7150.
Philpot BD, Cho KK, Bear MF (2007). Obligatory role of NR2A for metaplasticity in visual cortex. Neuron 53: 495–502.
Philpot BD, Espinosa JS, Bear MF (2003). Evidence for altered NMDA receptor function as a basis for metaplasticity in visual cortex. J Neurosci 23: 5583–5588.
Philpot BD, Sekhar AK, Shouval HZ, Bear MF (2001). Visual experience and deprivation bidirectionally modify the composition and function of NMDA receptors in visual cortex. Neuron 29: 157–169.
Piomelli D (2003). The molecular logic of endocannabinoid signalling. Nat Rev Neurosci 4: 873–884.
Pisani A, Centonze D, Bernardi G, Calabresi P (2005). Striatal synaptic plasticity: implications for motor learning and Parkinson's disease. Mov Disord 20: 395–402.
Pitler TA, Alger BE (1992). Postsynaptic spike firing reduces synaptic GABAA responses in hippocampal pyramidal cells. J Neurosci 12: 4122–4132.
Pocklington AJ, Cumiskey M, Armstrong JD, Grant SG (2006). The proteomes of neurotransmitter receptor complexes form modular networks with distributed functionality underlying plasticity and behaviour. Mol Syst Biol 2: 2006.0023.
Powell CM, Schoch S, Monteggia L, Barrot M, Matos MF, Feldmann N et al (2004). The presynaptic active zone protein RIM1alpha is critical for normal learning and memory. Neuron 42: 143–153.
Regehr WG, Tank DW (1991). The maintenance of LTP at hippocampal mossy fiber synapses is independent of sustained presynaptic calcium. Neuron 7: 451–459.
Reid CA, Dixon DB, Takahashi M, Bliss TV, Fine A (2004). Optical quantal analysis indicates that long-term potentiation at single hippocampal mossy fiber synapses is expressed through increased release probability, recruitment of new release sites, and activation of silent synapses. J Neurosci 24: 3618–3626.
Ressler KJ, Rothbaum BO, Tannenbaum L, Anderson P, Graap K, Zimand E et al (2004). Cognitive enhancers as adjuncts to psychotherapy: use of D-cycloserine in phobic individuals to facilitate extinction of fear. Arch Gen Psychiatry 61: 1136–1144. Evidence that an NMDA receptor agonist can enhance the efficacy of psychotherapy.
Reymann KG, Frey JU (2007). The late maintenance of hippocampal LTP: requirements, phases, ‘synaptic tagging’, ‘late-associativity’ and implications. Neuropharmacology 52: 24–40.
Robbe D, Alonso G, Chaumont S, Bockaert J, Manzoni OJ (2002a). Role of p/q-Ca2+ channels in metabotropic glutamate receptor 2/3-dependent presynaptic long-term depression at nucleus accumbens synapses. J Neurosci 22: 4346–4356.
Robbe D, Kopf M, Remaury A, Bockaert J, Manzoni OJ (2002b). Endogenous cannabinoids mediate long-term synaptic depression in the nucleus accumbens. Proc Natl Acad Sci USA 99: 8384–8388.
Rogan MT, Staubli UV, LeDoux JE (1997). Fear conditioning induces associative long-term potentiation in the amygdala. Nature 390: 604–607. Paper demonstrating that fear conditioning elicits LTP in the amygdala.
Rosahl TW, Geppert M, Spillane D, Herz J, Hammer RE, Malenka RC et al (1993). Short-term synaptic plasticity is altered in mice lacking synapsin I. Cell 75: 661–670.
Rosahl TW, Spillane D, Missler M, Herz J, Selig DK, Wolff JR et al (1995). Essential functions of synapsins I and II in synaptic vesicle regulation. Nature 375: 488–493.
Rubenstein JL, Merzenich MM (2003). Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behavior 2: 255–267.
Rumpel S, LeDoux J, Zador A, Malinow R (2005). Postsynaptic receptor trafficking underlying a form of associative learning. Science 308: 83–88. Evidence that learning induces synaptic AMPAR trafficking that is required for a form of associative memory.
Rush AM, Wu J, Rowan MJ, Anwyl R (2002). Group I metabotropic glutamate receptor (mGluR)-dependent long-term depression mediated via p38 mitogen-activated protein kinase is inhibited by previous high-frequency stimulation and activation of mGluRs and protein kinase C in the rat dentate gyrus in vitro. J Neurosci 22: 6121–6128.
Rutherford LC, Nelson SB, Turrigiano GG (1998). BDNF has opposite effects on the quantal amplitude of pyramidal neuron and interneuron excitatory synapses. Neuron 21: 521–530.
Saal D, Dong Y, Bonci A, Malenka RC (2003). Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron 37: 577–582. Experiments demonstrating that in vivo administration of drugs of abuse elicit ‘LTP’ at excitatory synapses on dopamine neurons.
Safo PK, Regehr WG (2005). Endocannabinoids control the induction of cerebellar LTD. Neuron 48: 647–659.
Sajikumar S, Navakkode S, Sacktor TC, Frey JU (2005). Synaptic tagging and cross-tagging: the role of protein kinase Mzeta in maintaining long-term potentiation but not long-term depression. J Neurosci 25: 5750–5756.
Salin PA, Malenka RC, Nicoll RA (1996). Cyclic AMP mediates a presynaptic form of LTP at cerebellar parallel fiber synapses. Neuron 16: 797–803.
Sanes JR, Lichtman JW (1999). Can molecules explain long-term potentiation? Nat Neurosci 2: 597–604.
Schluter OM, Xu W, Malenka RC (2006). Alternative N-terminal domains of PSD-95 and SAP97 govern activity-dependent regulation of synaptic AMPA receptor function. Neuron 51: 99–111.
Schmitz D, Mellor J, Breustedt J, Nicoll RA (2003). Presynaptic kainate receptors impart an associative property to hippocampal mossy fiber long-term potentiation. Nat Neurosci 6: 1058–1063.
Schnell E, Sizemore M, Karimzadegan S, Chen L, Bredt DS, Nicoll RA (2002). Direct interactions between PSD-95 and stargazin control synaptic AMPA receptor number. Proc Natl Acad Sci USA 99: 13902–13907.
Schuman EM, Dynes JL, Steward O (2006). Synaptic regulation of translation of dendritic mRNAs. J Neurosci 26: 7143–7146.
Selig DK, Hjelmstad GO, Herron C, Nicoll RA, Malenka RC (1995). Independent mechanisms for long-term depression of AMPA and NMDA responses. Neuron 15: 417–426.
Serrano P, Yao Y, Sacktor TC (2005). Persistent phosphorylation by protein kinase Mzeta maintains late-phase long-term potentiation. J Neurosci 25: 1979–1984.
Sheng M, Kim MJ (2002). Postsynaptic signaling and plasticity mechanisms. Science 298: 776–780.
Shi S, Hayashi Y, Esteban JA, Malinow R (2001). Subunit-specific rules governing AMPA receptor trafficking to synapses in hippocampal pyramidal neurons. Cell 105: 331–343.
Sigurdsson T, Doyere V, Cain CK, LeDoux JE (2007). Long-term potentiation in the amygdala: a cellular mechanism of fear learning and memory. Neuropharmacology 52: 215–227.
Silva AJ, Paylor R, Wehner JM, Tonegawa S (1992). Impaired spatial learning in alpha-calcium-calmodulin kinase II mutant mice. Science 257: 206–211.
Singla S, Kreitzer AC, Malenka RC (2007). Mechanisms for synapse specificity during striatal long-term depression. J Neurosci 27: 5260–5264.
Sjostrom PJ, Turrigiano GG, Nelson SB (2003). Neocortical LTD via coincident activation of presynaptic NMDA and cannabinoid receptors. Neuron 39: 641–654. Demonstration of a form of endocannabinoid-mediated LTD in neocortex.
Slepnev VI, Ochoa GC, Butler MH, Grabs D, De Camilli P (1998). Role of phosphorylation in regulation of the assembly of endocytic coat complexes. Science 281: 821–824.
Snyder EM, Colledge M, Crozier RA, Chen WS, Scott JD, Bear MF (2005). Role for A kinase-anchoring proteins (AKAPS) in glutamate receptor trafficking and long term synaptic depression. J Biol Chem 280: 16962–16968.
Snyder EM, Philpot BD, Huber KM, Dong X, Fallon JR, Bear MF (2001). Internalization of ionotropic glutamate receptors in response to mGluR activation. Nat Neurosci 4: 1079–1085.
Soderling TR, Derkach VA (2000). Postsynaptic protein phosphorylation and LTP. Trends Neurosci 23: 75–80.
Song I, Huganir RL (2002). Regulation of AMPA receptors during synaptic plasticity. Trends Neurosci 25: 578–588.
Spencer JP, Murphy KP (2002). Activation of cyclic AMP-dependent protein kinase is required for long-term enhancement at corticostriatal synapses in rats. Neurosci Lett 329: 217–221.
Spillane DM, Rosahl TW, Sudhof TC, Malenka RC (1995). Long-term potentiation in mice lacking synapsins. Neuropharmacology 34: 1573–1579.
Squire LR, Stark CE, Clark RE (2004). The medial temporal lobe. Annu Rev Neurosci 27: 279–306.
Stein V, House DR, Bredt DS, Nicoll RA (2003). Postsynaptic density-95 mimics and occludes hippocampal long-term potentiation and enhances long-term depression. J Neurosci 23: 5503–5506.
Steinberg JP, Takamiya K, Shen Y, Xia J, Rubio ME, Yu S et al (2006). Targeted in vivo mutations of the AMPA receptor subunit GluR2 and its interacting protein PICK1 eliminate cerebellar long-term depression. Neuron 49: 845–860.
Stellwagen D, Beattie EC, Seo JY, Malenka RC (2005). Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J Neurosci 25: 3219–3228.
Stellwagen D, Malenka RC (2006). Synaptic scaling mediated by glial TNF-alpha. Nature 440: 1054–1059.
Stent GS (1973). A physiological mechanism for Hebb's postulate of learning. Proc Natl Acad Sci USA 70: 997–1001.
Steward O, Schuman EM (2001). Protein synthesis at synaptic sites on dendrites. Annu Rev Neurosci 24: 299–325.
Sung KW, Choi S, Lovinger DM (2001). Activation of group I mGluRs is necessary for induction of long-term depression at striatal synapses. J Neurophysiol 86: 2405–2412.
Sutton MA, Schmidt EF, Choi KH, Schad CA, Whisler K, Simmons D et al (2003). Extinction-induced upregulation in AMPA receptors reduces cocaine-seeking behaviour. Nature 421: 70–75.
Sutton MA, Schuman EM (2006). Dendritic protein synthesis, synaptic plasticity, and memory. Cell 127: 49–58.
Sweatt JD (2004). Mitogen-activated protein kinases in synaptic plasticity and memory. Curr Opin Neurobiol 14: 311–317.
Takahashi T, Svoboda K, Malinow R (2003). Experience strengthening transmission by driving AMPA receptors into synapses. Science 299: 1585–1588.
Takumi Y, Ramirez-Leon V, Laake P, Rinvik E, Ottersen OP (1999). Different modes of expression of AMPA and NMDA receptors in hippocampal synapses. Nat Neurosci 2: 618–624.
Tamminga CA (1998). Schizophrenia and glutamatergic transmission. Crit Rev Neurobiol 12: 21–36.
Tang K, Low MJ, Grandy DK, Lovinger DM (2001). Dopamine-dependent synaptic plasticity in striatum during in vivo development. Proc Natl Acad Sci USA 98: 1255–1260.
Tang YP, Shimizu E, Dube GR, Rampon C, Kerchner GA, Zhuo M et al (1999). Genetic enhancement of learning and memory in mice. Nature 401: 63–69. Experiments showing that genetic manipulation of NMDAR subunit enhances both LTP and memory.
Thomas GM, Huganir RL (2004). MAPK cascade signalling and synaptic plasticity. Nat Rev Neurosci 5: 173–183.
Thomas MJ, Beurrier C, Bonci A, Malenka RC (2001). Long-term depression in the nucleus accumbens: a neural correlate of behavioral sensitization to cocaine. Nat Neurosci 4: 1217–1223.
Thomas MJ, Malenka RC (2003). Synaptic plasticity in the mesolimbic dopamine system. Philos Trans R S Lond 358: 815–819.
Thompson SM, Capogna M, Scanziani M (1993). Presynaptic inhibition in the hippocampus. Trends Neurosci 16: 222–227.
Tomita S, Adesnik H, Sekiguchi M, Zhang W, Wada K, Howe JR et al (2005a). Stargazin modulates AMPA receptor gating and trafficking by distinct domains. Nature 435: 1052–1058.
Tomita S, Stein V, Stocker TJ, Nicoll RA, Bredt DS (2005b). Bidirectional synaptic plasticity regulated by phosphorylation of stargazin-like TARPs. Neuron 45: 269–277.
Tsien JZ, Huerta PT, Tonegawa S (1996). The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell 87: 1327–1338.
Tsui J, Malenka RC (2006). Substrate localization creates specificity in calcium/calmodulin-dependent protein kinase II signaling at synapses. J Biol Chem 281: 13794–13804.
Tsvetkov E, Carlezon WA, Benes FM, Kandel ER, Bolshakov VY (2002). Fear conditioning occludes LTP-induced presynaptic enhancement of synaptic transmission in the cortical pathway to the lateral amygdala. Neuron 34: 289–300.
Turrigiano GG (2006). More than a sidekick: glia and homeostatic synaptic plasticity. Trends Mol Med 12: 458–460.
Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB (1998). Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature 391: 892–896. Demonstration of homeostatic synaptic scaling.
Turrigiano GG, Nelson SB (2004). Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci 5: 97–107.
Ungless MA, Whistler JL, Malenka RC, Bonci A (2001). Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature 411: 583–587.
Vanderschuren LJ, Kalivas PW (2000). Alterations in dopaminergic and glutamatergic transmission in the induction and expression of behavioral sensitization: a critical review of preclinical studies. Psychopharmacology 151: 99–120.
Vekovischeva OY, Zamanillo D, Echenko O, Seppala T, Uusi-Oukari M, Honkanen A et al (2001). Morphine-induced dependence and sensitization are altered in mice deficient in AMPA-type glutamate receptor-A subunits. J Neurosci 21: 4451–4459.
Walker DL, Ressler KJ, Lu KT, Davis M (2002). Facilitation of conditioned fear extinction by systemic administration or intra-amygdala infusions of D-cycloserine as assessed with fear-potentiated startle in rats. J Neurosci 22: 2343–2351.
Wang H, Wagner JJ (1999). Priming-induced shift in synaptic plasticity in the rat hippocampus. J Neurophysiol 82: 2024–2028.
Wang YT, Linden DJ (2000). Expression of cerebellar long-term depression requires postsynaptic clathrin-mediated endocytosis. Neuron 25: 635–647.
Wang Z, Kai L, Day M, Ronesi J, Yin HH, Ding J et al (2006). Dopaminergic control of corticostriatal long-term synaptic depression in medium spiny neurons is mediated by cholinergic interneurons. Neuron 50: 443–452.
Watt AJ, van Rossum MC, MacLeod KM, Nelson SB, Turrigiano GG (2000). Activity coregulates quantal AMPA and NMDA currents at neocortical synapses. Neuron 26: 659–670.
Wenthold RJ, Petralia RS, Blahos II J, Niedzielski AS (1996). Evidence for multiple AMPA receptor complexes in hippocampal CA1/CA2 neurons. J Neurosci 16: 1982–1989.
Whitlock JR, Heynen AJ, Shuler MG, Bear MF (2006). Learning induces long-term potentiation in the hippocampus. Science 313: 1093–1097. Evidence that learning can induce LTP in the hippocampus.
Wilson RI, Nicoll RA (2002). Endocannabinoid signaling in the brain. Science 296: 678–682.
Wolf ME (1998). The role of excitatory amino acids in behavioral sensitization to psychomotor stimulants. Prog Neurobiol 54: 679–720.
Xia J, Chung HJ, Wihler C, Huganir RL, Linden DJ (2000). Cerebellar long-term depression requires PKC-regulated interactions between GluR2/3 and PDZ domain-containing proteins. Neuron 28: 499–510.
Xiao MY, Zhou Q, Nicoll RA (2001). Metabotropic glutamate receptor activation causes a rapid redistribution of AMPA receptors. Neuropharmacology 41: 664–671.
Xu J, Kang J (2005). The mechanisms and functions of activity-dependent long-term potentiation of intrinsic excitability. Rev Neurosci 16: 311–323.
Young JZ, Isiegas C, Abel T, Nguyen PV (2006). Metaplasticity of the late-phase of long-term potentiation: a critical role for protein kinase A in synaptic tagging. Eur J Neurosci 23: 1784–1794.
Yuste R, Bonhoeffer T (2001). Morphological changes in dendritic spines associated with long-term synaptic plasticity. Annu Rev Neurosci 24: 1071–1089.
Zakharenko SS, Zablow L, Siegelbaum SA (2002). Altered presynaptic vesicle release and cycling during mGluR-dependent LTD. Neuron 35: 1099–1110.
Zalutsky RA, Nicoll RA (1990). Comparison of two forms of long-term potentiation in single hippocampal neurons. Science 248: 1619–1624. Experiments defining the mechanisms of presynaptic LTP at hippocampal mossy fiber synapses.
Zhang W, Linden DJ (2003). The other side of the engram: experience-driven changes in neuronal intrinsic excitability. Nat Rev Neurosci 4: 885–900.
Zhou Q, Homma KJ, Poo MM (2004). Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron 44: 749–757.
Zhou Y, Wu H, Li S, Chen Q, Cheng XW, Zheng J et al (2006). Requirement of TORC1 for Late-Phase Long-Term Potentiation in the Hippocampus. PLoS ONE 1: E16.
Zhu JJ, Qin Y, Zhao M, Van Aelst L, Malinow R (2002). Ras and Rap control AMPA receptor trafficking during synaptic plasticity. Cell 110: 443–455.
Zhu Y, Pak D, Qin Y, McCormack SG, Kim MJ, Baumgart JP et al (2005). Rap2-JNK removes synaptic AMPA receptors during depotentiation. Neuron 46: 905–916.
Ziff EB (2007). TARPs and the AMPA receptor trafficking paradox. Neuron 53: 627–633.
Zilberter Y (2000). Dendritic release of glutamate suppresses synaptic inhibition of pyramidal neurons in rat neocortex. J Physiol 528: 489–496.
Zilberter Y, Kaiser KM, Sakmann B (1999). Dendritic GABA release depresses excitatory transmission between layer 2/3 pyramidal and bitufted neurons in rat neocortex. Neuron 24: 979–988.
Zola-Morgan S, Squire LR (1993). Neuroanatomy of memory. Annu Rev Neurosci 16: 547–563.
Zucker RS, Regehr WG (2002). Short-term synaptic plasticity. Annu Rev Physiol 64: 355–405.
Acknowledgements
RM gratefully acknowledges support from NIMH, NIDA, NINDS, and NARSAD. AC acknowledges the support of the The International Human Frontier Science Program Organization. We apologize to those colleagues whose work was not cited because of the large scope of this review and severe space limitations.
Author information
Authors and Affiliations
Corresponding author
Additional information
DISCLOSURE/CONFLICT OF INTEREST
RM is on the board of scientific advisors for Merck Inc., Renovis Inc. and Seaside Therapeutics. These duties had no influence on the content of this article. AC has no disclosures or conflict of interest.
Rights and permissions
About this article
Cite this article
Citri, A., Malenka, R. Synaptic Plasticity: Multiple Forms, Functions, and Mechanisms. Neuropsychopharmacol 33, 18–41 (2008). https://doi.org/10.1038/sj.npp.1301559
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.npp.1301559
Keywords
This article is cited by
-
From high-throughput transcriptome characterization of individual synaptosomes to constructing the whole-brain connectome
Neuropsychopharmacology (2024)
-
The amygdala NT3-TrkC pathway underlies inter-individual differences in fear extinction and related synaptic plasticity
Molecular Psychiatry (2024)
-
d-Cycloserine enhances the bidirectional range of NMDAR-dependent hippocampal synaptic plasticity
Translational Psychiatry (2024)
-
Astrocytes in the adult dentate gyrus—balance between adult and developmental tasks
Molecular Psychiatry (2024)
-
Alpha-Asarone Ameliorates Neurological Dysfunction of Subarachnoid Hemorrhagic Rats in Both Acute and Recovery Phases via Regulating the CaMKII-Dependent Pathways
Translational Stroke Research (2024)
