
Abstract
Blockade of brain μ-opioid receptor (μ-OR) and δ-opioid receptor (δ-OR) was investigated in recently abstinent alcohol-dependent subjects (N=21) maintained on naltrexone. Subjects completed a 19-day inpatient protocol, which included alcohol abstinence followed by naltrexone treatment (50 mg) on days 15–19. Blood samples were collected after the first administration of naltrexone to evaluate serum levels of naltrexone and 6-β-naltrexol. Regional brain μ-OR binding potential (BP) and δ-OR Ki was measured using [11C]carfentanil (CAR) positron emission tomography (PET) and [11C]methyl naltrindole ([11C]MeNTI) PET, respectively, before (day 5) and during naltrexone treatment (day 18). Naltrexone inhibition of [11C]CAR BP was near maximal across all brain regions of interest with little variability across subjects (mean+SD% inhibition=94.9+4.9%). Naltrexone only partially inhibited the [11C]MeNTI Ki and there was more variability across subjects (mean+SD% inhibition=21.1+14.49%). Peak serum levels of naltrexone were positively correlated with % inhibition of δ-OR Ki in neocortex and basal ganglia. Peak serum levels of naltrexone were not correlated with % inhibition of μ-OR BP. Peak levels of 6-β-naltrexol were not significantly correlated with % inhibition of μ-OR BP or δ-OR Ki. Thus, the FDA recommended therapeutic dose of naltrexone was sufficient to produce near complete inhibition of the μ-OR in recently abstinent alcohol dependent subjects. The lower percent inhibition of δ-OR and greater variability in δ-OR blockade by naltrexone across subjects may contribute to individual differences in treatment outcomes to naltrexone. Further investigations on the relationship between individual differences in δ-OR blockade by naltrexone and clinical outcomes should be explored.
Similar content being viewed by others
INTRODUCTION
There is strong evidence supporting an association between alcoholism and the endogenous opioid system (for reviews see Gianoulakis, 2004; Oswald and Wand, 2004). There are three major opioid peptides, β-endorphin, enkephalins, and dynorphins, which target μ, δ, and κ subtypes of the opioid receptors (ORs). β-Endorphin binds with equal affinity to μ- and δ-OR subtypes. Enkephalins bind to both μ- and δ-OR subtypes, but the affinity to δ-OR subtypes is about 20-fold greater. In contrast, dynorphins are relatively selective for κ-ORs. It has been proposed that μ, δ, and κ subtypes of the ORs modulate different subjective effects of alcohol. Specifically, the μ- and δ-ORs are thought to be important for alcohol reinforcement and the maintenance of alcohol consumption, whereas activation of κ-ORs is thought to modulate alcohol's dysphoric effects.
Consistent with the proposed role of μ- and δ-ORs in modulating alcohol reinforcement and consumption, numerous studies have demonstrated that alcohol preferring rodent strains show differences in μ- and δ-OR density (de Waele et al, 1995; Marinelli et al, 2000; McBride et al, 1998; Soini et al, 1998) and basal levels of β-endorphin and enkephalins (Blum et al, 1987; De Waele and Gianoulakis, 1994; De Waele et al, 1992; Gianoulakis et al, 1992; Nylander et al, 1994) when compared with nonpreferring strains. Similarly, μ-OR knockout mice self-administer alcohol at lower levels when compared with wild-type controls (Becker et al, 2002; Hall et al, 2001; Roberts et al, 2000). In rodents, chronic alcohol consumption has been shown to increase μ-OR binding in limbic areas including the nucleus accumbens (Cowen et al, 1998, 1999; Djouma and Lawrence, 2002). Only two studies have examined μ-OR binding in alcohol dependent subjects. One study found that the availability of μ-ORs in the ventral striatum, including the nucleus accumbens, was higher in recently abstinent alcohol-dependent subjects when compared with healthy control subjects (Heinz et al, 2005). A second study (Bencherif et al, 2004), reported mean μ-OR binding potential (BP) for alcohol-dependent subjects was lower in the right dorsal lateral prefrontal cortex, the right anterior frontal cortex, and right parietal cortex, when compared with normal healthy control subjects. The nucleus accumbens was not analyzed.
The role of μ- and δ-ORs in alcohol consumption and reinforcement is further supported by results of preclinical studies in which the opioid system is perturbed via pharmacological manipulations. Naloxone, naltrexone, and nalmefene are nonselective OR antagonists in that they bind at μ, δ, and κ subtypes of the receptors. Administration of these nonselective OR antagonists decreased alcohol consumption in both rodents (Froehlich et al, 1990; Hubbell et al, 1986; Samson and Doyle, 1985; Weiss et al, 1990) and monkeys (Altshuler et al, 1980; Boyle et al, 1998; Kornet et al, 1991; Myers et al, 1986; Williams et al, 1998). Conversely, administration of opioid agonists (eg morphine) increased alcohol intake in rodents (Czirr et al, 1987; Hubbell et al, 1986; Linseman and Harding, 1990; Nichols et al, 1991; Reid et al, 1986). Antagonists that are selective for the μ-ORs (Froehlich et al, 1991; Krishnan-Sarin et al, 1998) or δ-ORs also can decrease alcohol drinking (Franck et al, 1998; Froehlich et al, 1991; June et al, 1999; Krishnan-Sarin et al, 1995a, 1995b) (but cf. (Honkanen et al, 1996; Hyytia, 1993; Middaugh et al, 2000; Stromberg et al, 1998; Williams and Woods, 1998)). Thus, the role of the specific OR subtypes in mediating alcohol consumption and reinforcement is still under investigation.
Naltrexone is a Federal Drug Agency (FDA) approved medication for treatment of alcohol dependence. Two important double-blind placebo-controlled clinical trials (O'Malley et al, 1992; Volpicelli et al, 1992) first demonstrated that, when combined with psychosocial treatment, the nonselective OR antagonist naltrexone reduced craving and the number of alcohol-drinking days in recently abstinent alcoholics. Since then, numerous clinical studies have been conducted. Recent reviews and meta-analyses of these randomized clinical trials indicate that naltrexone is effective in reducing drinking and relapse, however, not all individuals show improvement (Anton and Swift, 2003; Garbutt et al, 1999; Mann, 2004). The optimal dosing regimen, duration of treatment, and identification of individual patient characteristics that predict a successful outcome with naltrexone administration are still under investigation.
The present study was conducted to examine μ- and δ-OR availability in recently abstinent alcoholics before and during treatment with the FDA recommended therapeutic naltrexone dose of 50 mg/day p.o. Subjects were enrolled in a 19-day inpatient study and μ- and δ-OR availability before and during naltrexone treatment were determined using [11C]carfentanil (CAR) and [11C]methyl maltrindole ([11C]MeNTI) positron emission tomography (PET), respectively. The inter- and intra-subject variabilities in μ- and δ-OR availability across different brain regions during naltrexone treatment were characterized. In addition, serum levels of naltrexone and its biologically active metabolite 6-β-naltrexol were determined.
METHODS
Subjects
Subjects were recruited via advertisement and provided informed consent(s), in the sober state, using an Institutional Review Board-approved informed consent document. They were then interviewed using a battery of diagnostic and psychological instruments. Subjects met DSM-IV criteria for alcohol dependence based on the Semi-Structured Assessment of the Genetics of Alcoholism (Bucholz et al, 1994), and were actively drinking at hazardous levels (ie consuming 60 or more drinks/month and at least 5 drinks/occasion weekly prior to hospitalization) as determined by completion of a 90-day Time Line Follow Back of drinking (Sobell and Sobell, 1992). Alcohol dependence was further characterized using the Alcohol Dependence Scale (ADS) (Skinner and Allen, 1982). Subjects also completed the Fagerstom Nicotine Dependence Test (FNDT) to determine the level of nicotine dependence associated with smoking. Individuals were excluded if they met current DSM-IV diagnostic criteria for any other Axis-I disorder, including drug abuse/dependence (except nicotine), if urine drug toxicology was positive at screening or hospital admission, or if they had other ongoing health problems. Subjects were also screened for prior alcohol withdrawal symptoms and were excluded if they reported alcohol-related seizures or the need for medication during previous detoxifications. Additional exclusion criteria were in effect once subjects enrolled in the inpatient protocol; subjects were excluded if the medically supervised withdrawal was severe (eg Clinical Institute Withdrawal Assessment (CIWA) scores 32 or more), and/or if subjects required medication (eg benzodiazepines) to alleviate withdrawal. Fifteen alcohol-dependent men and six alcohol-dependent women (total N=21) were enrolled in the study and admitted to the Johns Hopkins Hospital General Clinical Research Center (GCRC) for 19 days. Demographic characteristics, nicotine dependence status, drinking status, and assessment data for the subjects are shown in Table 1.
General Procedures
Following hospital admission, subjects completed medically supervised alcohol withdrawal. Subjects completed the Clinical Institute Withdrawal Assessment-Alcohol Revised (CIWA-Ar) (Sullivan et al, 1989), 3–4 times each day for the first 5 days. Subjects were instructed to mark items to reflect the time period since the last measurement. No subject required withdrawal medication based on CIWA scores, vital signs and physician assessment. Subjects could not smoke during their hospitalization. In order to reduce potential confounds introduced by different levels of smoking and/or effects of nicotine withdrawal, we used a standardized approach to medicate all nicotine-dependent subjects with transdermal nicotine patches (21 mg nicotine) each day and maintained this dose throughout the inpatient protocol. During hospitalization, subjects had random urine tests and breath alcohol tests to verify abstinence from alcohol and drugs. Subjects received naltrexone (50 mg, p.o.) at 0900 and 2100 hours on day 15, and then at 2100 each day for the remainder of the study. Naltrexone was administered under nurse supervision; subjects were observed to verify that the pill was swallowed to ensure medication compliance. Subjects completed a daily questionnaire to detect and rate severity of various symptoms such as sleep disturbances, nervousness, anxiety, gastrointestinal symptoms (cramps, nausea, vomiting, constipation), decreased appetite, low energy, increased energy, dizziness and headache. Side effects of naltrexone appeared to be mild, as there was no systematic change in self-reported symptoms (eg nausea, gastrointestinal upset, restlessness, fatigue) during naltrexone treatment as compared with those reported during the inpatient stay prior to naltrexone induction. None of the subjects required a change in dose or discontinuation of the medication.
Naltrexone and 6-β-Naltrexol Sampling Procedures
On day 15, blood samples were collected in 16 of the subjects before and after administration of the first dose of naltrexone to evaluate serum levels of naltrexone and 6-β-naltrexol. Five of the subjects either declined to participate or there were technical difficulties in blood collection for this aspect of the study. In order to control for possible effects of food intake on drug absorption, all subjects received a calorie-controlled breakfast at 0730. An intravenous catheter was placed in the nondominant forearm at 0800 (ie 1 h before naltrexone was administered). After 2 baseline samples, (−15 min and 0), the naltrexone capsule was swallowed, and then serum samples were collected at 30-min intervals for 4 h.
Levels of naltrexone and 6-β-naltrexol in K3-EDTA-treated human plasma were determined by gas chromatography/tandem mass spectrometry by Cedra Corporation (Austin, TX; analytical test method #870). Briefly, plasma-containing naltrexone, 6-β-naltrexol, and the internal standards, naloxone and 6-β-naltrexol-D3, was extracted with an organic solvent mixture under alkaline conditions. Following centrifugation, the upper organic layer was removed and further cleansed by back-extraction. The dried extract was reconstituted. An aliquot of the extract was injected onto a SCIEX API 4000 LC-MS-MS equipped with an HPLC column. The peak area of the m/z 342 → 324 naltrexone product ion was measured against the peak area of the m/z 328 → 310 naloxone internal standard product ion. The peak area of the m/z 344 → 326 6-β-naltrexol product ion is measured against the peak area of the m/z 347 → 329 6-β-naltrexol-D3 internal standard product ion. Quantitation was performed using weighted linear least squares regression analyses generated from fortified plasma calibration standards prepared immediately prior to each run. The range of quantitation was 0.0200–2.00 ng/ml for naltrexone and 0.0200–4.00 ng/ml for 6-β-naltrexol based on the analysis of 0.500 ml of plasma.
PET Imaging Procedures
During the inpatient stay, subjects underwent 2 days of PET imaging. All 21 subjects completed PET scans for μ-OR availability using [11C]CAR. Fifteen of the 21 subjects also completed scans for δ-OR using [11C]MeNTI; two subjects were unable to complete the naltrindole scans due to problems with tracer synthesis on scheduled PET days and four subjects refused the arterial line placement, which is required only for the naltrindole scan. Before admission, subjects were fitted with a thermoplastic mask that was individually fitted to each subject's face for immobilization during imaging.
Approximately 1 week before admission, all subjects underwent magnetic resonance imaging (MRI) to allow anatomical localization and alignment of PET imaging planes within and across subjects (Meltzer et al, 1990). Pre-naltrexone PET imaging was conducted immediately after alcohol withdrawal symptoms subsided (day 5). PET imaging during naltrexone maintenance was conducted after subjects had received four doses of 50 mg (day 18) in order to target when naltrexone and 6-β-naltrexol serum levels were stable. In our previous study (McCaul et al, 2000b), levels of naltrexone and 6-β-naltrexol were very stable over consecutive days of administration of 50 mg naltrexone, with very low variability within individual subjects.
Subjects underwent PET imaging using [11C]CAR, a specific μ-OR agonist (Frost et al, 1985; Titeler et al, 1989), and [11C]MeNTI, a specific δ-OR agonist, developed for PET imaging (Lever et al, 1992; Madar et al, 1996). On each day, PET imaging was conducted; there was a fixed order for the two scans; the [11C]MeNTI scan was initiated at 0830 followed by the [11C]CAR scan at 1045. We maintained a fixed order of scans to assure that both scans could be conducted on the same day. The [11C]MeNTI scan was conducted first to avoid potential problems related to possible pharmacological effects of [11C]CAR. [11C]CAR can produce OR agonistic effects, in spite of using a sub-therapeutic dose, whereas [11C]MeNTI does not. In addition, since [11C]CAR and [11C]MeNTI have specific binding affinity to different subtypes of opioid receptors ([11C]CAR to μ-OR, and [11C]MeNTI to δ-OR), binding of [11C]MeNTI radiotracer would not affect binding for [11C]CAR. All subjects received a calorie-controlled breakfast 3 h before the first scan. For nicotine-dependent subjects, nicotine transdermal patches were applied 3 h before the [11C]MeNTI scan.
PET Image Acquisition
PET scans were acquired in 3D mode on a GE Advance PET scanner (GE Medical Systems, Milwaukee, WI). A transmission scan of 10-min duration was obtained using rotating germanium-68 rods before injection of the radiotracer. After intravenous bolus administration of the radiotracers [11C]CAR (19.4±2.1 mCi SA: 17 298±13 907 mCi/μmol) for [11C]MeNTI and (19.2±3.2 mCi SA: 4447.5±2960.7 mCi/μmol), a set of 25 images with variable time period (6 × 30 s, 5 × 60 s, 5 × 120 s, 9 × 480 s) was acquired during a 90-min period for each study. After correction for attenuation using the transmission scan, images were reconstructed in a 128 × 128 × 35 matrix with pixel size of 2 × 2 × 4.25 mm with filtered back projection methods using a ramp filter and decay-correction.
PET Image Analysis
Parametric images of the distribution volume ratio (DVR) for [11C]CAR were generated by voxel-based Logan noninvasive graphical analysis (Endres et al, 2003; Logan et al, 1996) using the occipital cortex as a reference region or region with negligible receptors (Endres et al, 2003). The K2 of the reference region (occipital cortex) equals 0.104 min−1 (K2R) as published previously (Endres et al, 2003). Distribution volume images were converted to BP images by subtracting one from all voxels to be proportional to Bmax/Kd.
Parametric images of the input constant (Ki) of [11C]MeNTI were generated by Patlak graphical analysis (Patlak and Blasberg, 1985; Smith et al, 1999) using metabolite-corrected plasma radioactivity as the input function (See below). We selected this approach because our previous study demonstrated that dissociation of the ligand was not evident for 90 min post injection (Madar et al, 1996; Smith et al, 1999).
The proportion of metabolized [11C]MeNTI in plasma was determined from arterial blood samples obtained before injection and at 5, 10, 15, 30, 45, 60, 75, and 90 min after injection. The first four samples were 8 ml, and the remaining were 16 ml. These samples were centrifuged at 2000 r.p.m. for 5 min, the plasma removed and passed through an activated C18 reverse-phase Sep-Pak (Waters Associates, Milford, MA, USA). The Sep-Pak was washed with 0.1 mmol/l ammonium formate and eluted with 100% methanol. The methanol fraction was diluted with an aqueous solution of 2% triethylamine and 3% acetic acid to a final solution of 40% methanol. This solution was passed through a C18 reverse-phase analytical HPLC column (Waters Associates). The HPLC mobile phase was composed of 1:1 acetonitrile and methanol (40%) and HPLC-grade water buffered with 2% triethylamine and 3% acetic acid (60%). The HPLC flow rate was set at 3 ml/min. Extraction efficiencies for the parent compound and radiolabeled metabolites were calculated and corrected for at each step of the Sep-Pak separation. The pre-injection blood sample, with [11C]MeNTI added, was used as a control to determine whether metabolites were produced in vitro and the extraction efficiency of the parent drug. A sample of blood obtained 90 min after tracer administration, with high metabolite content, was used to calculate the metabolite extraction fraction. The total concentration of radioactivity in plasma then was corrected for the presence of radiolabeled metabolites by multiplying the plasma concentration by the fraction of unchanged drug using linear interpolation. Further details on these procedures can be found in prior publications (Frost et al, 1989; Price et al, 1993; Sadzot et al, 1991). Briefly, the HPLC system was equipped with two opposed 12.7-cm (5-inch) NaI (T1) detectors, a strip chart recorder, and a computer interface. Each radioactive peak was automatically integrated and the decay corrected back to the time of sample injection into the column. The percentage of total activity corresponding to unmetabolized [11C]MeNTI was then calculated for each time point.
The parametric images of [11C]CAR and [11C]MeNTI were spatially normalized into the standard space of the Montreal Neurological Institute (MNI). First, 0–90 min average images of each scan were spatially normalized to the ligand-specific template in MNI space. Then, the transformation function, obtained from 30–90 min average images, was applied to the parametric images. Standard regions of interests (ROIs) for several brain regions were applied to the spatially normalized parametric images.
Data Analysis
The mean values of normalized μ-OR BP and δ-OR Ki (net input) were determined before and during naltrexone administration and were compared using paired t-tests with Bonferroni adjustment for distribution of error (p<0.05/total number of comparisons); a p-value of less than 0.003 was accepted as significant for each pair-wise comparison. To determine if basic demographic variables (race, gender, age), nicotine dependence status as defined by score on the Fagerstrom Nicotine Dependence Test, drinking status (years of dependent drinking) or ADS score were correlated with percent inhibition of μ-OR and δ-OR by naltrexone, data were analyzed using SAS (version 9.1) CORR procedure. The results of these analyses without Bonferroni adjustment are presented in the results section. We then used the conservative statistical approach to include variables that showed a correlation of 0.5 or greater with a p<0.05 as covariates in subsequent analyses. The relationship of peak serum levels of naltrexone and 6-β-naltrexol to percent change in μ-OR BP and δ-OR Ki during naltrexone administration were analyzed using SAS (version 9.1) CORR procedure and controlling for current nicotine dependence status.
RESULTS
μ-OR Binding before and during Naltrexone
Figure 1 shows mean [11C]CAR PET images of brain μ-OR before and during naltrexone treatment in 21 alcohol dependent subjects. As shown in Figure 1 (top panel), scans obtained before naltrexone treatment revealed high [11C]CAR BP in the frontal cortex, temporal cortex, amygdala, thalamus, and pituitary gland. Intermediate [11C]CAR BP is seen in the anterior cingulate, and putamen and very low BP is seen in occipital cortex and the primary motor cortex. In contrast, PET images conducted during naltrexone treatment (Figure 1, bottom panels) revealed that naltrexone completely inhibited [11C]CAR BP across all brain regions, with the exception of some non-specific binding in the pituitary gland.

Mean images of the distribution of μ-OR in the brain of 21 alcoholics after IV administration of [11C]CAR during scans conducted pre-naltrexone treatment (top panel) and during naltrexone treatment (bottom panel). Images shown are color-coded according to the scale shown (0–1.5) so that highest concentrations of the radiotracer are represented by red and lowest concentrations by black/purple.
Table 2 shows [11C]CAR BP, using ROI methodology, before and during naltrexone treatment. Before naltrexone treatment, [11C]CAR BP was high across all ROIs, but was greatest in caudate, putamen, amygdala, and thalamus. Naltrexone decreased [11C]CAR BP across all ROIs. Paired t-test comparisons of μ-OR BP before and during naltrexone revealed a significant (p<0.0001) difference in means across all ROIs (Table 2).
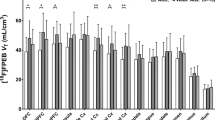
Figure 2 shows the percent decrease from baseline in [11C]CAR BP during naltrexone treatment. Naltrexone inhibition of [11C]CAR BP was near maximal across all brain ROIs in recently abstinent, alcohol-dependent subjects. When compared with the pre-naltrexone baseline, the mean (+SD) percent decrease in [11C]CAR BP across all brain ROIs was 94.9 (+4.9%). In addition to naltrexone producing maximal inhibition of [11C]CAR BP across brain areas, there was very low variability in the percent inhibition of [11C]CAR BP across subjects for each ROI.

Changes in μ-OR BP during treatment with naltrexone. Data are shown as percent decrease from baseline ((Basal−inhibition)/Basal × 100) across brain regions of interest (ROIs). Bars are group means and data points represent individual subjects.
δ-OR Binding before and during Naltrexone
Figure 3 shows PET images of brain δ-OR in 15 alcohol dependent subjects and Table 3 shows [11C]MeNTI Ki across the ROIs before and during naltrexone treatment. As shown in the top panel of Figure 3, scans conducted before naltrexone treatment revealed intermediate [11C]MeNTI Ki across brain ROIs, except for thalamus and cerebellum where Ki was lower. Naltrexone inhibited [11C]MeNTI Ki across brain ROIs, but [11C]MeNTI Ki was still low to intermediate (Figure 3, bottom panel). Paired t-test comparisons of [11C]MeNTI Ki before and during naltrexone treatment revealed a significant (p<0.003) mean difference across all ROIs except for the thalamus and cerebellum, where changes were not significant (Table 3).

Mean images of Patlak slope of δ-OR in the brain of 15 alcoholics after IV administration of [11C]MeNTI during scans conducted pre-naltrexone treatment (top panel) and during naltrexone treatment (bottom panel). Images shown are color-coded according to the scale shown (0–0.1) so that highest concentrations of the radiotracer are represented by red and lowest concentrations by black/purple.
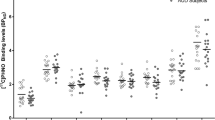
Figure 4 shows the percent decrease from baseline in [11C]MeNTI Ki during naltrexone treatment. [11C]MeNTI Ki was only partially inhibited by naltrexone across all brain ROIs (mean+SD% inhibition=21.1+14.49%), and was highly variable across subjects for each region. For example, some individuals showed a 50% decrease in [11C]MeNTI Ki, whereas others showed slight increases or decreases in [11C]MeNTI Ki from pre-naltrexone levels.

Changes in δ-OR Ki following treatment with naltrexone. Data are shown as percent decrease from baseline ((Basal−inhibition)/Basal × 100) across brain regions of interest (ROIs). Bars are group means and data points represent individual subjects.
Demographics and Behavioral Variables
Analysis of demographic variables indicated that race, gender and age were not correlated with percent decrease in μ-OR BP or δ-OR Ki during naltrexone treatment. Nicotine dependence status was positively correlated with percent decrease in δ-OR Ki during naltrexone treatment in the inferior temporal cortex (0.57, p<0.03), medial frontal cortex (0.54, p<0.04) and midoccipital cortex (0.56, p<0.03). That is, nicotine-dependent subjects evidenced greater decreases in δ-OR Ki. In contrast, nicotine dependence status was not correlated with percent change in μ-OR BP during naltrexone treatment. Table 4 shows percent decrease in μ-OR BP and δ-OR Ki in nicotine dependent and nondependent subjects. Years of dependent drinking were positively correlated with percent change in μ-OR BP during naltrexone treatment in the anterior cingulate (0.48, p<0.03). The number of years of dependent drinking was not correlated with percent decrease in δ-OR Ki during naltrexone treatment. ADS score was not correlated with percent decrease in μ-OR BP or δ-OR Ki during naltrexone treatment.
Effects of Peak Serum Levels of Naltrexone and 6-β-Naltrexol on μ-OR and δ-OR Binding
Mean peak serum levels of naltrexone and 6-β-naltrexol were 16.13 (+9.37 SD) ng/ml and 73.31 (+23.3 SD) ng/ml, respectively. Analysis of serum samples collected on the first day of naltrexone administration revealed substantial individual differences in peak serum levels of both naltrexone (range 4.7–33.2, Figure 5a) and 6-β-naltrexol (range 34.5–114.1, Figure 5b). Similarly, the metabolism of naltrexone to 6-β-naltrexol was widely variable across subjects; at peak levels in serum, the naltrexone to 6-β-naltrexol ratio ranged from 1.3 to 14.1 across subjects (mean 6.1).

Peak serum levels (ng/ml) of naltrexone and 6-β-naltrexol in 16 abstinent alcoholic subjects following oral administration of 50 mg naltrexone. Data shown are the peak serum level for (a) naltrexone and (b) 6-β-naltrexol determined on the first day of naltrexone administration; data points represent individual subjects and the horizontal line is the group mean.
When controlling for nicotine dependence status, analysis of the 14 subjects who completed both [11C]CAR PET and the blood sampling protocol indicated that peak serum levels of naltrexone were not correlated with percent decrease in [11C]CAR BP during naltrexone administration. When controlling for nicotine dependence status, analysis of the 11 subjects who completed both [11C]MeNTI PET and the blood sampling protocols indicated that peak serum levels of naltrexone were positively correlated with percent change in [11C]MeNTI Ki during naltrexone in caudate (0.76, p<0.02), putamen (0.67, p<0.04), cingulate (0.64, p<0.05), insular cortex (0.73, p<0.02), inferior parietal cortex (0.69, p<0.03), midfrontal cortex (0.66, p<0.04) and superior parietal cortex (0.65, p<0.04). Peak levels of 6-β-naltrexol were not significantly correlated with changes in [11C]CAR BP or [11C]MeNTI Ki during naltrexone administration.
DISCUSSION
The major finding of this study was that the standard dose of 50 mg p.o. naltrexone was sufficient to produce near complete inhibition of the μ-OR in recently abstinent alcohol dependent subjects, but only partial inhibition of the δ-OR. Although there was marked inter-subject variability in serum levels of naltrexone and 6-β-naltrexol, there was very little inter-subject variability of μ-OR availability within each brain ROI.
The high level of μ-OR inhibition is important as the μ-OR is thought to be the principal site of action for naltrexone's therapeutic effects for alcohol dependence (Herz, 1997). Meta-analyses of randomized clinical trials have demonstrated that 50 mg naltrexone has an overall small-to-moderate effect size in reducing drinking and relapse in alcohol dependent subjects (Anton and Swift, 2003; Garbutt et al, 1999; Mann, 2004). Unfortunately, not all individuals show improvement during naltrexone treatment at the FDA-recommended dose of 50 mg. The source of variability in naltrexone's therapeutic efficacy is still under investigation, but several possibilities have been proposed. These include poor medication compliance, incomplete receptor blockade due to differences in naltrexone metabolism, or incomplete receptor blockade due to differences in μ-OR affinity or number in alcoholics (McCaul et al, 2000b). Another possible source of variability is differences in opioid neurotransmitter system sensitivity reported in individuals with family histories of alcoholism (Wand et al, 2001, 1999).
In the current study, δ-OR Ki at baseline was highest in neocortical regions (insular, parietal, frontal, cingulate, and occipital), caudate nucleus, and putamen, and lowest in the cerebellum and thalamus. The brain regions of high binding determined with Patlak slope in the current study are consistent with those determined in our previous studies (Madar et al, 1996) in which [11C]MeNTI normalized binding to δ-OR receptors was calculated using the cerebellum as a reference region (ROI minus cerebellum/cerebellum). Patlak graphical analysis was selected for use in the current study based on our findings in our previous study (Smith et al, 1999) that evaluated both kinetic (two- and three-compartment models, Patlak graphical analysis) and nonkinetic (apparent volume of distribution and activity at a late scanning time) analytic approaches. It was determined that Patlak graphical analyses yielded less-biased binding-related measures compared with the nonkinetic methods (eg distribution volume). In addition, [11C]MeNTI shows irreversible binding characteristics during the 90-min scan period (Madar et al, 1996).
In the current study, treatment with 50 mg naltrexone resulted in a partial (mean: 21%) inhibition of δ-OR Ki across ROIs when compared with the pre-naltrexone baseline. Interestingly, the magnitude of inhibition of δ-OR Ki by naltrexone was highly variable across subjects; some subjects showed an almost 50% decrease in δ-OR Ki, but others showed only slight decreases or even slight increases. The essentially complete blocking of μ-OR and less significant blocking of δ-OR with the same dose of naltrexone is consistent with the affinity of naltrexone for the different OR subtypes. Although naltrexone binds at μ, δ, and κ subtypes of the OR (ie is a nonselective OR antagonist), it binds with higher affinity at the μ-OR (Wang et al, 2001). Thus, the greater inhibition of μ-OR vs δ-OR by 50 mg naltrexone is likely related to higher affinity of naltrexone binding at μ-OR. Since binding at δ- and κ-OR increases as the naltrexone dose is increased (Wang et al, 2001), we hypothesize that δ-OR blockade by naltrexone would likely be greater if subjects were treated with higher doses of naltrexone (eg 100 mg).
Previous studies have shown that μ-OR BP (Bmax/Kd) increases with age in neocortical areas and the putamen (Zubieta et al, 1999). In addition, sex differences in μ-OR BP in a number of cortical and subcortical areas have been reported, with higher μ-OR binding in women (Zubieta et al, 1999). The lower level of δ-OR Ki compared with μ-OR BP during naltrexone administration and the variability in magnitude of inhibition in the current study does not appear to be related to gender, age, race, or severity of alcohol dependence (as determined by ADS score). None of these variables was correlated with percent decrease in μ-OR or δ-OR binding.
Naltrexone undergoes first-pass metabolism in the liver to its major metabolite 6-β-naltrexol. 6-β-naltrexol is biologically active and has a longer half-life (12–18 h) than naltrexone (4–9 h) (Davidson et al, 1996; Ferrari et al, 1998; Verebey et al, 1976; Wall et al, 1981). In the current study, there was marked variability across subjects for serum levels of naltrexone, and there was a fourfold difference in peak serum levels of naltrexone vs 6-β-naltrexol levels. These data are consistent with naltrexone metabolism previously reported in heavy drinkers and in opioid dependent subjects (McCaul et al, 2000b; Verebey et al, 1976). It has been suggested that increasing the dose of naltrexone to increase levels of naltrexone and 6-β-naltrexol in blood may be needed to achieve successful treatment in some individuals (McCaul et al, 2000a; Rohsenow, 2004). Previous studies in our laboratory have shown that doubling the dose of naltrexone from 50 to 100 mg was more effective in reducing subjective effects of alcohol (ie ratings of alcohol ‘liking’ and ‘best effects’) in heavy drinkers and also doubled the serum levels of 6-β-naltrexol (McCaul et al, 2000b). However, it is unknown what serum levels of naltrexone and 6-β-naltrexol are needed to achieve adequate occupancy at the δ-OR in the brain for therapeutic effectiveness. Medication compliance is important as Volpicelli and colleagues have shown that naltrexone was effective only in subjects with 90% or greater medication compliance (Volpicelli et al, 1997). In the current study, medication compliance was carefully controlled. Naltrexone was administered under nurse supervision and subjects were observed to verify that each dose was taken. Thus, medication compliance did not contribute to individual variability in naltrexone blockade of the δ-OR.
Smoking and nicotine dependence status may be a factor in the variability found in δ-OR binding. There is strong evidence of interactions between nicotinic and opioid mechanisms with regard to nicotine dependence (McGehee, 2006; Pomerleau, 1998). In humans, endogenous opioid levels increase following smoking (Pomerleau et al, 1983). Similarly, systemic administration of nicotine increases the release of endogenous opioids in the nucleus accumbens and striatum in rodents (Davenport et al, 1990; Dhatt et al, 1995; Houdi et al, 1991; Walters et al, 2005). Chronic nicotine has been shown to increase μ-OR in striatum in female rats and in the ventral tegmental area in male and female mice (Walters et al, 2005; Wewers et al, 1999). The opiate antagonist naloxone can block the subjective effects of nicotine in chronic smokers (Brauer et al, 1999; King and Meyer, 2000). Thus, it is not surprising that naltrexone has been proposed for the treatment of nicotine dependence (Schnoll and Lerman, 2006). In the current study, there was no relationship between nicotine dependence status and change in μ-OR availability, but there was a modest relationship between nicotine dependence status and δ-OR blockade by naltrexone in three brain regions during naltrexone treatment. Specifically, δ-OR blockade by naltrexone was higher in individuals who had higher Fagerstrom scores of nicotine dependence in inferior temporal cortex, medial frontal cortex and midoccipital cortex. The lack of significant correlations for μ-OR blockade is not surprising since naltrexone produced near maximal blockade of μ-OR with low variability across subjects (ie a ceiling effect). Although the relationship between nicotine dependence and naltrexone therapeutic efficacy was not the focus of the current study, this is clearly an important research area for future studies.
Since there was a correlation between Fagerstrom nicotine dependence score and the magnitude of δ-OR blockade by naltrexone, nicotine dependence status was included as a control variable in the analyses of the effects of serum levels of naltrexone and 6-β-naltrexol on δ-OR blockade. When controlling for nicotine dependence status, serum naltrexone levels were positively correlated with δ-OR blockade in neocortical and basal ganglia regions. That is, the magnitude of δ-OR blockade was higher in individuals who had higher peak serum levels of naltrexone. These data suggest that individual differences in OR activity as well as naltrexone bioavailability and metabolism may be important for naltrexone's therapeutic efficacy to the extent that the δ-OR is involved in modulating alcohol reinforcement. Future data analyses will examine further the possible influence of nicotine use and dependence on OR availability, and on naltrexone bioavailability, metabolism, and elimination rates.
There were numerous procedural strengths to the study design. Since subjects were required to remain on the GCRC for the duration of the study, alcohol abstinence, and medication compliance were verified. This provided the opportunity to repeatedly collect the PET images and to safely avoid the possible confound of medicated alcohol withdrawal. In particular, subjects did not receive any sedative drugs (eg benzodiazepines), which may alter OR function (Cox and Collins, 2001). There were also some study limitations that may limit the generalization of these findings. These include the small sample sizes, selection of alcohol-dependent subjects without serious withdrawal symptoms, possible effects of nicotine replacement therapy, and potential differences associated with PET scan order and test-retest reproducibility. It is also possible there may be changes in the δ-OR during immediate withdrawal and throughout the course of long-term alcohol abstinence. Thus, the 2-week abstinence period between the baseline and naltrexone PET scans may have had an impact on these results, however, these procedures closely parallel the clinical practice. Heinz et al (2005) showed similar μ-OR BP in 5-week abstinent alcohol dependent subjects as our 5-day-abstinent subjects.
In summary, these data demonstrate that the standard clinical dose of 50 mg p.o. naltrexone was sufficient to produce near complete inhibition of the μ-OR in recently abstinent alcohol-dependent subjects, and that, μ-OR binding potential was very similar across all subjects. In contrast, naltrexone produced only partial inhibition of the δ-OR and variability across subjects was high. Based on these findings, the use of higher naltrexone doses does not appear necessary to block μ-OR during early abstinence. However, higher doses of naltrexone have been shown to attenuate the subjective effects of alcohol (McCaul et al, 2000b), and may also be needed to increase blockade of δ-OR. Thus, we hypothesize that the lower percent inhibition of δ-OR and greater variability in δ-OR blockade by naltrexone across subjects may contribute to individual differences in treatment outcomes to naltrexone. Subjects in this study will be further evaluated in follow-up visits and continued naltrexone treatment. Once this next phase of the study is completed, it will be possible to examine the specific relationship between μ- and δ-OR blockade by naltrexone and treatment outcomes to naltrexone.
References
Altshuler HL, Phillips PE, Feinhandler DA (1980). Alteration of ethanol self-administration by naltrexone. Life Sci 26: 679–688.
Anton RF, Swift RM (2003). Current pharmacotherapies of alcoholism: a US perspective. Am J Addict 12 (Suppl 1): S53–S68.
Becker A, Grecksch G, Kraus J, Loh HH, Schroeder H, Hollt V (2002). Rewarding effects of ethanol and cocaine in mu opioid receptor-deficient mice. Naunyn Schmiedebergs Arch Pharmacol 365: 296–302.
Bencherif B, Wand GS, McCaul ME, Kim YK, Ilgin N, Dannals RF et al (2004). Mu-opioid receptor binding measured by [11C]carfentanil positron emission tomography is related to craving and mood in alcohol dependence. Biol Psychiatry 55: 255–262.
Blum K, Wallace JE, Hall CW, Trachtenberg MA, Briggs AH (1987). Regional brain [Met]-enkephalin in alcohol-preferring and non-alcohol-preferring inbred strains of mice. Experientia 43: 408–410.
Boyle AE, Stewart RB, Macenski MJ, Spiga R, Johnson BA, Meisch RA (1998). Effects of acute and chronic doses of naltrexone on ethanol self- administration in rhesus monkeys. Alcohol Clin Exp Res 22: 359–366.
Brauer LH, Behm FM, Westman EC, Patel P, Rose JE (1999). Naltrexone blockade of nicotine effects in cigarette smokers. Psychopharmacology (Berl) 143: 339–346.
Bucholz KK, Cadoret R, Cloninger CR, Dinwiddie SH, Hesselbrock VM, Nurnberger Jr JI et al (1994). A new, semi-structured psychiatric interview for use in genetic linkage studies: a report on the reliability of the SSAGA. J Stud Alcohol 55: 149–158.
Cowen MS, Rezvani A, Jarrott B, Lawrence AJ (1998). Distribution of opioid peptide gene expression in the limbic system of Fawn-Hooded (alcohol-preferring) and Wistar-Kyoto (alcohol-non-preferring) rats. Brain Res 796: 323–326.
Cowen MS, Rezvani AH, Jarrott B, Lawrence AJ (1999). Ethanol consumption by Fawn-Hooded rats following abstinence: effect of naltrexone and changes in mu-opioid receptor density. Alcohol Clin Exp Res 23: 1008–1014.
Cox RF, Collins MA (2001). The effects of benzodiazepines on human opioid receptor binding and function. Anesth Analg 93: 354–358, 353rd contents page.
Czirr SA, Hubbell CL, Milano WC, Frank JM, Reid LD (1987). Selected opioids modify intake of sweetened ethanol solution among female rats. Alcohol 4: 157–160.
Davenport KE, Houdi AA, Van Loon GR (1990). Nicotine protects against mu-opioid receptor antagonism by beta-funaltrexamine: evidence for nicotine-induced release of endogenous opioids in brain. Neurosci Lett 113: 40–46.
Davidson AF, Emm TA, Pieniaszek Jr HJ (1996). Determination of naltrexone and its major metabolite, 6-beta-naltrexol, in human plasma using liquid chromatography with electrochemical detection. J Pharm Biomed Anal 14: 1717–1725.
de Waele JP, Gianoulakis C (1994). Enhanced activity of the brain beta-endorphin system by free-choice ethanol drinking in C57BL/6 but not DBA/2 mice. Eur J Pharmacol 258: 119–129.
de Waele JP, Kiianmaa K, Gianoulakis C (1995). Distribution of the mu and delta opioid binding sites in the brain of the alcohol-preferring AA and alcohol-avoiding ANA lines of rats. J Pharmacol Exp Ther 275: 518–527.
de Waele JP, Papachristou DN, Gianoulakis C (1992). The alcohol-preferring C57BL/6 mice present an enhanced sensitivity of the hypothalamic beta-endorphin system to ethanol than the alcohol-avoiding DBA/2 mice. J Pharmacol Exp Ther 261: 788–794.
Dhatt RK, Gudehithlu KP, Wemlinger TA, Tejwani GA, Neff NH, Hadjiconstantinou M (1995). Preproenkephalin mRNA and methionine-enkephalin content are increased in mouse striatum after treatment with nicotine. J Neurochem 64: 1878–1883.
Djouma E, Lawrence AJ (2002). The effect of chronic ethanol consumption and withdrawal on mu-opioid and dopamine D(1) and D(2) receptor density in Fawn-Hooded rat brain. J Pharmacol Exp Ther 302: 551–559.
Endres CJ, Bencherif B, Hilton J, Madar I, Frost JJ (2003). Quantification of brain mu-opioid receptors with [11C]carfentanil: reference-tissue methods. Nucl Med Biol 30: 177–186.
Ferrari A, Bertolotti M, Dell'Utri A, Avico U, Sternieri E (1998). Serum time course of naltrexone and 6 beta-naltrexol levels during long-term treatment in drug addicts. Drug Alcohol Depend 52: 211–220.
Franck J, Lindholm S, Raaschou P (1998). Modulation of volitional ethanol intake in the rat by central delta- opioid receptors. Alcohol Clin Exp Res 22: 1185–1189.
Froehlich JC, Harts J, Lumeng L, Li TK (1990). Naloxone attenuates voluntary ethanol intake in rats selectively bred for high ethanol preference. Pharmacol Biochem Behav 35: 385–390.
Froehlich JC, Zweifel M, Harts J, Lumeng L, Li TK (1991). Importance of delta opioid receptors in maintaining high alcohol drinking. Psychopharmacology 103: 467–472.
Frost JJ, Douglass KH, Mayberg HS, Dannals RF, Links JM, Wilson AA et al (1989). Multicompartmental analysis of [11C]-carfentanil binding to opiate receptors in humans measured by positron emission tomography. J Cereb Blood Flow Metab 9: 398–409.
Frost JJ, Wagner Jr HN, Dannals RF, Ravert HT, Links JM, Wilson AA et al (1985). Imaging opiate receptors in the human brain by positron tomography. J Comput Assist Tomogr 9: 231–236.
Garbutt JC, West SL, Carey TS, Lohr KN, Crews FT (1999). Pharmacological treatment of alcohol dependence: a review of the evidence. JAMA 281: 1318–1325.
Gianoulakis C (2004). Endogenous opioids and addiction to alcohol and other drugs of abuse. Curr Top Med Chem 4: 39–50.
Gianoulakis C, de Waele JP, Kiianmaa K (1992). Differences in the brain and pituitary beta-endorphin system between the alcohol-preferring AA and alcohol-avoiding ANA rats. Alcohol Clin Exp Res 16: 453–459.
Hall FS, Sora I, Uhl GR (2001). Ethanol consumption and reward are decreased in mu-opiate receptor knockout mice. Psychopharmacology (Berl) 154: 43–49.
Heinz A, Reimold M, Wrase J, Hermann D, Croissant B, Mundle G et al (2005). Correlation of stable elevations in striatal mu-opioid receptor availability in detoxified alcoholic patients with alcohol craving: a positron emission tomography study using carbon 11-labeled carfentanil. Arch Gen Psychiatry 62: 57–64.
Herz A (1997). Endogenous opioid systems and alcohol addiction. Psychopharmacology (Berl) 129: 99–111.
Honkanen A, Vilamo L, Wegelius K, Sarviharju M, Hyytia P, Korpi ER (1996). Alcohol drinking is reduced by a mu 1- but not by a delta-opioid receptor antagonist in alcohol-preferring rats. Eur J Pharmacol 304: 7–13.
Houdi AA, Pierzchala K, Marson L, Palkovits M, Van Loon GR (1991). Nicotine-induced alteration in Tyr-Gly-Gly and Met-enkephalin in discrete brain nuclei reflects altered enkephalin neuron activity. Peptides 12: 161–166.
Hubbell CL, Czirr SA, Hunter GA, Beaman CM, LeCann NC, Reid LD (1986). Consumption of ethanol solution is potentiated by morphine and attenuated by naloxone persistently across repeated daily administrations. Alcohol 3: 39–54.
Hyytia P (1993). Involvement of mu-opioid receptors in alcohol drinking by alcohol-preferring AA rats. Pharmacol Biochem Behav 45: 697–701.
June HL, McCane SR, Zink RW, Portoghese PS, Li TK, Froehlich JC (1999). The delta 2-opioid receptor antagonist naltriben reduces motivated responding for ethanol. Psychopharmacology (Berl) 147: 81–89.
King AC, Meyer PJ (2000). Naltrexone alteration of acute smoking response in nicotine-dependent subjects. Pharmacol Biochem Behav 66: 563–572.
Kornet M, Goosen C, Van Ree JM (1991). Effect of naltrexone on alcohol consumption during chronic alcohol drinking and after a period of imposed abstinence in free-choice drinking rhesus monkeys. Psychopharmacology 104: 367–376.
Krishnan-Sarin S, Jing SL, Kurtz DL, Zweifel M, Portoghese PS, Li TK et al (1995a). The delta opioid receptor antagonist naltrindole attenuates both alcohol and saccharin intake in rats selectively bred for alcohol preference. Psychopharmacology (Berl) 120: 177–185.
Krishnan-Sarin S, Portoghese PS, Li TK, Froehlich JC (1995b). The delta 2-opioid receptor antagonist naltriben selectively attenuates alcohol intake in rats bred for alcohol preference. Pharmacol Biochem Behav 52: 153–159.
Krishnan-Sarin S, Wand GS, Li XW, Portoghese PS, Froehlich JC (1998). Effect of mu opioid receptor blockade on alcohol intake in rats bred for high alcohol drinking. Pharmacol Biochem Behav 59: 627–635.
Lever JR, Scheffel U, Kinter CM, Ravert HT, Dannals RF, Wagner Jr HN et al (1992). In vivo binding of N1'-([11C]methyl)naltrindole to delta-opioid receptors in mouse brain. Eur J Pharmacol 216: 459–460.
Linseman MA, Harding S (1990). Intracerebroventricular morphine enhances alcohol consumption by rats. Pharmacol Biochem Behav 36: 405–408.
Logan J, Fowler JS, Volkow ND, Wang GJ, Ding YS, Alexoff DL (1996). Distribution volume ratios without blood sampling from graphical analysis of PET data. J Cereb Blood Flow Metab 16: 834–840.
Madar I, Lever JR, Kinter CM, Scheffel U, Ravert HT, Musachio JL et al (1996). Imaging of delta opioid receptors in human brain by N1′- ([11C]methyl)naltrindole and PET. Synapse 24: 19–28.
Mann K (2004). Pharmacotherapy of alcohol dependence: a review of the clinical data. CNS Drugs 18: 485–504.
Marinelli PW, Kiianmaa K, Gianoulakis C (2000). Opioid propeptide mRNA content and receptor density in the brains of AA and ANA rats. Life Sci 66: 1915–1927.
McBride WJ, Chernet E, McKinzie DL, Lumeng L, Li TK (1998). Quantitative autoradiography of mu-opioid receptors in the CNS of alcohol-naive alcohol-preferring P and -nonpreferring NP rats. Alcohol 16: 317–323.
McCaul ME, Wand GS, Eissenberg T, Rohde CA, Cheskin LJ (2000a). Naltrexone alters subjective and psychomotor responses to alcohol in heavy drinking subjects. Neuropsychopharmacology 22: 480–492.
McCaul ME, Wand GS, Rohde C, Lee SM (2000b). Serum 6-beta-naltrexol levels are related to alcohol responses in heavy drinkers. Alcohol Clin Exp Res 24: 1385–1391.
McGehee DS (2006). Nicotinic and opioid receptor interactions in nicotine addiction. Mol Interv 6: 311–314.
Meltzer CC, Bryan RN, Holcomb HH, Kimball AW, Mayberg HS, Sadzot B et al (1990). Anatomical localization for PET using MR imaging. J Comput Assist Tomogr 14: 418–426.
Middaugh LD, Kelley BM, Groseclose CH, Cuison Jr ER (2000). Delta-opioid and 5-HT3 receptor antagonist effects on ethanol reward and discrimination in C57BL/6 mice. Pharmacol Biochem Behav 65: 145–154.
Myers RD, Borg S, Mossberg R (1986). Antagonism by naltrexone of voluntary alcohol selection in the chronically drinking macaque monkey. Alcohol 3: 383–388.
Nichols ML, Hubbell CL, Kalsher MJ, Reid LD (1991). Morphine increases intake of beer among rats. Alcohol 8: 237–240.
Nylander I, Hyytia P, Forsander O, Terenius L (1994). Differences between alcohol-preferring (AA) and alcohol-avoiding (ANA) rats in the prodynorphin and proenkephalin systems. Alcohol Clin Exp Res 18: 1272–1279.
O'Malley SS, Jaffe AJ, Chang G, Schottenfeld RS, Meyer RE, Rounsaville B (1992). Naltrexone and coping skills therapy for alcohol dependence. A controlled study. Arch Gen Psychiatry 49: 881–887.
Oswald LM, Wand GS (2004). Opioids and alcoholism. Physiol Behav 81: 339–358.
Patlak CS, Blasberg RG (1985). Graphical evaluation of blood-to-brain transfer constants from multiple-time uptake data. Generalizations. J Cereb Blood Flow Metab 5: 584–590.
Pomerleau OF (1998). Endogenous opioids and smoking: a review of progress and problems. Psychoneuroendocrinology 23: 115–130.
Pomerleau OF, Fertig JB, Seyler LE, Jaffe J (1983). Neuroendocrine reactivity to nicotine in smokers. Psychopharmacology (Berl) 81: 61–67.
Price JC, Mayberg HS, Dannals RF, Wilson AA, Ravert HT, Sadzot B et al (1993). Measurement of benzodiazepine receptor number and affinity in humans using tracer kinetic modeling, positron emission tomography, and [11C]flumazenil. J Cereb Blood Flow Metab 13: 656–667.
Reid LD, Czirr SA, Milano WC, Hubbell CL, Manha NA (1986). Opioids and intake of alcoholic beverages. NIDA Res Monogr 75: 359–362.
Roberts AJ, McDonald JS, Heyser CJ, Kieffer BL, Matthes HW, Koob GF et al (2000). mu-Opioid receptor knockout mice do not self-administer alcohol. J Pharmacol Exp Ther 293: 1002–1008.
Rohsenow DJ (2004). What place does naltrexone have in the treatment of alcoholism? CNS Drugs 18: 547–560.
Sadzot B, Price JC, Mayberg HS, Douglass KH, Dannals RF, Lever JR et al (1991). Quantification of human opiate receptor concentration and affinity using high and low specific activity [11C]diprenorphine and positron emission tomography. J Cereb Blood Flow Metab 11: 204–219.
Samson HH, Doyle TF (1985). Oral ethanol self-administration in the rat: effect of naloxone. Pharmacol Biochem Behav 22: 91–99.
Schnoll RA, Lerman C (2006). Current and emerging pharmacotherapies for treating tobacco dependence. Expert Opin Emerg Drugs 11: 429–444.
Skinner HA, Allen BA (1982). Alcohol dependence syndrome: measurement and validation. J Abnorm Psychol 91: 199–209.
Smith JS, Zubieta JK, Price JC, Flesher JE, Madar I, Lever JR et al (1999). Quantification of delta-opioid receptors in human brain with N1'-([11C]methyl) naltrindole and positron emission tomography. J Cereb Blood Flow Metab 19: 956–966.
Sobell LC, Sobell MB (1992). Timeline followback: a technique for assessing self-reported alcohol consumption. In: Litten RZ, Allen J (eds). Measuring Alcohol Consumption: Psychosocial And Biological Methods. Humana Press: New Jersey. pp 41–72.
Soini SL, Ovaska T, Honkanen A, Hyytia P, Korpi ER (1998). Brain opioid receptor binding of [3H]CTOP and [3H]naltrindole in alcohol-preferring AA and alcohol-avoiding ANA rats. Alcohol 15: 227–232.
Stromberg MF, Casale M, Volpicelli L, Volpicelli JR, O'Brien CP (1998). A comparison of the effects of the opioid antagonists naltrexone, naltrindole, and beta-funaltrexamine on ethanol consumption in the rat. Alcohol 15: 281–289.
Sullivan JT, Sykora K, Schneiderman J, Naranjo CA, Sellers EM (1989). Assessment of alcohol withdrawal: the revised clinical institute withdrawal assessment for alcohol scale (CIWA-Ar). Br J Addict 84: 1353–1357.
Titeler M, Lyon RA, Kuhar MJ, Frost JF, Dannals RF, Leonhardt S et al (1989). Mu opiate receptors are selectively labelled by [3H]carfentanil in human and rat brain. Eur J Pharmacol 167: 221–228.
Verebey K, Volavka J, Mule SJ, Resnick RB (1976). Naltrexone: disposition, metabolism, and effects after acute and chronic dosing. Clin Pharmacol Ther 20: 315–328.
Volpicelli JR, Alterman AI, Hayashida M, O'Brien CP (1992). Naltrexone in the treatment of alcohol dependence. Arch Gen Psychiatry 49: 876–880.
Volpicelli JR, Rhines KC, Rhines JS, Volpicelli LA, Alterman AI, O'Brien CP (1997). Naltrexone and alcohol dependence. Role of subject compliance. Arch Gen Psychiatry 54: 737–742.
Wall ME, Brine DR, Perez-Reyes M (1981). The metabolism of naltrexone in man. NIDA Res Monogr 28: 105–131.
Walters CL, Cleck JN, Kuo YC, Blendy JA (2005). Mu-opioid receptor and CREB activation are required for nicotine reward. Neuron 46: 933–943.
Wand G, McCaul ME, Gotjen D, Reynolds J, Lee S (2001). Confirmation that offspring from families with alcohol-dependent individuals have greater hypothalamic-pituitary-adrenal axis activation induced by naloxone compared with offspring without a family history of alcohol dependence. Alcohol Clin Exp Res 25: 1134–1139.
Wand GS, Mangold D, Ali M, Giggey P (1999). Adrenocortical responses and family history of alcoholism. Alcohol Clin Exp Res 23: 1185–1190.
Wang D, Raehal KM, Bilsky EJ, Sadee W (2001). Inverse agonists and neutral antagonists at mu opioid receptor (MOR): possible role of basal receptor signaling in narcotic dependence. J Neurochem 77: 1590–1600.
Weiss F, Mitchiner M, Bloom FE, Koob GF (1990). Free-choice responding for ethanol versus water in alcohol preferring (P) and unselected Wistar rats is differentially modified by naloxone, bromocriptine, and methysergide. Psychopharmacology 101: 178–186.
Wewers ME, Dhatt RK, Snively TA, Tejwani GA (1999). The effect of chronic administration of nicotine on antinociception, opioid receptor binding and met-enkelphalin levels in rats. Brain Res 822: 107–113.
Williams KL, Winger G, Pakarinen ED, Woods JH (1998). Naltrexone reduces ethanol- and sucrose-reinforced responding in rhesus monkeys. Psychopharmacology (Berl) 139: 53–61.
Williams KL, Woods JH (1998). Oral ethanol-reinforced responding in rhesus monkeys: effects of opioid antagonists selective for the mu-, kappa-, or delta-receptor. Alcohol Clin Exp Res 22: 1634–1639.
Zubieta JK, Dannals RF, Frost JJ (1999). Gender and age influences on human brain mu-opioid receptor binding measured by PET. Am J Psychiatry 156: 842–848.
Acknowledgements
This research was supported by the National Institute of Alcohol Abuse and Alcoholism (AA11872, AA11855, and AA12303). We acknowledge the technical support of Dr Hayden T Ravert, Mr Robert Smoot, and Mr Daniel Holt for their radiochemistry expertise, and Ms Karen Edmonds and David Clough for their PET acquisition and reconstruction expertise. We also thank Dr Hiroto Kuwabara for helpful comments and suggestions on this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclosure/Conflict of Interest
The author(s) declare that, except for income received from the primary employer, no financial support or compensation has been received from any individual or corporate entity over the past 3 years for research or professional service and there are no personal financial holdings that could be perceived as constituting a potential conflict of interest.
Rights and permissions
About this article
Cite this article
Weerts, E., Kim, Y., Wand, G. et al. Differences in δ- and μ-Opioid Receptor Blockade Measured by Positron Emission Tomography in Naltrexone-Treated Recently Abstinent Alcohol-Dependent Subjects. Neuropsychopharmacol 33, 653–665 (2008). https://doi.org/10.1038/sj.npp.1301440
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.npp.1301440
Keywords
This article is cited by
-
Opioid antagonism in humans: a primer on optimal dose and timing for central mu-opioid receptor blockade
Neuropsychopharmacology (2023)
-
The Utilization of Low Dose Naltrexone for Chronic Pain
CNS Drugs (2023)
-
Impaired TRPM3-dependent calcium influx and restoration using Naltrexone in natural killer cells of myalgic encephalomyelitis/chronic fatigue syndrome patients
Journal of Translational Medicine (2022)
-
Effects of low-dose naltrexone on quality of life in high-grade glioma patients: a placebo-controlled, double-blind randomized trial
Supportive Care in Cancer (2022)
-
The changing opioid crisis: development, challenges and opportunities
Molecular Psychiatry (2021)
