
Abstract
We originally suggested that inhibition of tumor necrosis factor-related apoptosis inducing ligand (TRAIL) death pathway could be taken into consideration as a potential therapeutic strategy for Alzheimer's disease (AD). However, because the critical role of TRAIL in immune surveillance, the neutralization of TRAIL protein by an antibody to prevent its binding to death receptors is definitely a risky approach. Here, we demonstrated that the blockade of the TRAIL death receptor DR5 with a specific antibody completely prevented amyloid β peptide (Aβ) neurotoxicity in both neuronal cell line and primary cortical neurons. DR5 was demonstrated to be a key factor in TRAIL death pathway. In fact, whereas TRAIL expression was enhanced dose-dependently by concentrations of β amyloid ranging from 10 nM to 1 μM, only the highest toxic dose of Aβ (25 μM) induced the increased expression of DR5 and neuronal cell death. In addition, the increased expression of DR5 receptor after β amyloid treatment was sustained by p53 transcriptional activity, as demonstrated by the data showing that the p53 inhibitor Pifithrin α prevented both β amyloid-induced DR5 induction and cell death. These data suggest a sequential activation of p53 and DR5 upon β amyloid exposure. Further insight into the key role of DR5 in AD was suggested by data showing a significant increase of DR5 receptor in cortical slices of AD brain. Thus, these findings may give intracellular TRAIL pathway a role in AD pathophysiology, making DR5 receptor a possible candidate as a pharmacological target.
Similar content being viewed by others


INTRODUCTION
One of the more accredited candidates involved in etiopathology of Alzheimer's disease (AD) is thought to be the amyloid β peptide (Aβ), which is the major component of the extracellular deposition named senile plaques (Mori et al, 1992; LeVine, 2004; Heppner et al, 2004). Senile plaques, together with intracellular depositions of neurofibrillary tangle, are the two anatomo-pathological hall markers that characterize this disease. Aβ is a 39–43 amino-acid length peptide derived from the amyloidogenic processing of the larger amyloid precursor protein (APP) (Haas et al, 1992; Seubert et al, 1992). The central role of Aβ in AD is sustained by the discovery of APP mutation in some familiar cases of the disease (Selkoe, 1996; Kowalska, 2003). This mutations have been demonstrated to be associated with an elevated production of Aβ peptide 1–40 and 1–42, which have the tendency to aggregate to form final unsoluble fibrils (Dewachter et al, 2000; Lewis et al, 2004; Helpern et al, 2004). The result coming from genetic studies and in vitro and in vivo experiments on animal models of AD gives Aβ a neurotoxic effect (Cotman et al, 1992; Gray and Patel, 1995; Oster-Granite et al, 1996; Moechars et al, 1999; Ekinci et al, 2000; Peng et al, 2002; Tamagno et al, 2003; Rosales-Corral et al, 2004).
It is well demonstrated that Aβ causes different intracellular effects, involving different pathways that can proceed either separately or sequentially. We have recently shown that one of the members of the TNF (tumor necrosis factor)-α family, the TNF-related apoptosis inducing ligand (TRAIL), contributes substantially to amyloid-induced neurotoxicity in human SH-SY5Y neuronal cell line (Cantarella et al, 2003). TRAIL is a novel peptide molecule belonging to the TNF family, whose main role is to induce programmed cell death in tumor cell lines from various tissue origins (Pitti et al, 1996). TRAIL binds to five specific receptors of the TNF/NGF family (Pan et al, 1997a; Walczak et al, 1997). However, only two of them, DR4 and DR5, have an intracellular death domain, which appears essential for triggering cell death processes (Pan et al, 1997a; Walczak et al, 1997). Although the decoy receptors TRAIL-R3 and -R4 act as a dominant negative of TRAIL proapoptic effects (Pan et al, 1997b; Degli-Esposti et al, 1997; Sheridan et al, 1997), a fifth receptor, named osteoprotegerin, appears to be involved in bone remodelling (Emery et al, 1998).
The contribution of TRAIL to Aβ neurotoxicity was demonstrated by data showing that anti-TRAIL-neutralizing monoclonal antibody protects neuronal SH-SY5Y cells from Aβ neurotoxicity (Cantarella et al, 2003). Furthermore, we recently showed TRAIL-positive cells in the brain of AD patients. TRAIL staining was localized in the cerebral cortex of human brain slices, often in the proximity of Congo Red-positive amyloid plaques (Uberti et al, 2004). Thus, these findings may give intracellular TRAIL pathway a role in AD pathophysiology, and indicate TRAIL peptide as a suitable target for pharmacological intervention. However, given the key role of TRAIL in providing and maintaining immune surveillance, questions have been raised regarding the potential usefulness of an antibody anti-TRAIL in AD therapy. Thus, we addressed our study toward another possible candidate belonging to the TRAIL pathway, the TRAIL DR5 receptor. Interestingly enough, TRAIL DR5 receptor is weakly expressed in human brain, and it is not detectable in other human organs (Ichikawa et al, 2001). In contrast, many human cancer tissues reacted positively to anti-DR5 antibody. In the brain, DR5 receptor is substantially expressed in neurons and oligodendrocytes, suggesting that the exposure to TRAIL could potentially induce apoptosis in these cells (Dorr et al, 2002). Consistent with this hypothesis, recombinant TRAIL was found to induce apoptosis in human neuroblastoma cell line (Milani et al, 2003), rat cortical neurons and in human brain slices (Nitsch et al, 2000). In this study, we investigated the involvement of TRAIL death receptor DR5 in Aβ-mediated apoptosis in both human neuronal cell line and mouse cortical neurons. Furthermore, we characterized the expression of the DR5 receptor in human brain derived from an AD case.
MATERIALS AND METHODS
Cell Culture
Neuroblastoma cell line SH-SY5Y was routinely cultured in 1:1 Ham's F12:Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% (v/v) fetal calf serum, 2 mM glutamine, 50 μg/ml penicillin, and 100 μg/ml streptomycin and was kept at 37°C in a humidified 5% CO2/95% air. For differentiation, cultures were seeded at approximately 105 cells/dish and retinoic acid was added to a final concentration of 10 μM. The medium was changed every day and cultures were allowed to differentiate for 2 weeks.
Primary Cultures of Mouse Cortical Neurons
Fifteen embryonic day mice were taken with Caesarean section from anesthetized pregnant dams. C57/BL6 mice were purchased from Charles River, Italy. Cerebral cortices were isolated and dissociated by manual dispersion with a fire-polished Pasteur pipette. Cells were plated at a density of 1.5 × 105 cells/cm2. Culture dishes were coated with 10 μg/ml poly-L-lysine. The cells were plated in Neurobasal medium (Invitrogen Corporation) supplemented with 2% B27 (Invitrogen Corporation), 0.5 mM L-glutamine, and 50 U/ml penicillin/streptomycin (Invitrogen Corporation). Three days after plating, 50% of the medium was changed with fresh medium and subsequently 50% of the medium was changed twice a week, until 11 days in vitro.
Human Tissues
Brain tissues were removed for diagnostic purposes during autopsy from three AD patients and from one cognitive normal, age-matched individual deceased owing to non-neurological disease. One AD case was a female died at the age of 78 years with a 5-year history of sporadic AD. The other two cases were siblings (II-3, age of death 65 years, disease duration 7 years; II-5, age of death 56 years, disease duration 6 years) affected by a familiar form of AD caused by PS2 M239I mutation (Zatti et al, 2004). The neuropathological diagnosis of AD was performed in accordance with CERAD recommendations (Mirra et al, 1991) by the local neuropathology unit at the Department of Pathology.
Handling of β-Amyloid Peptide
Aβ1–42 and Aβ25–35 were purchased from Bachem Feinchemikalien AG (Bubendorf, Switzerland). Different lots of the peptides were dissolved in sterile, doubly distilled water at a concentration of 2.5 mM and stored at −20°C. Aβ1–42 stock solutions were kept for 1 week in a 37°C incubator so as to allow aggregation and, therefore, toxicity. Aβ25–35 was used immediately after suspension (Pike et al, 1993). All peptides were tested at graded concentrations (range: 10 nM–10 μM) for evaluation of cell toxicity.
Evaluation of Cell Viability
5 × 103 cells/well were plated in 96-well plates and grown to 70–80% confluence in complete DMEM containing 10 μM retinoic acid. Differentiated cultures were incubated for 48 h with 25 μM βAP25–35, or βAP1–42 in DMEM with 1% serum at 37°C. These agents were performed alone or in combination with Pifithrin α (PIF) (200 nM), an inhibiting DR5 antibody (1 μg/ml; Alexis Biochemicals, San Diego, CA) or an isotype-matched control (IgG1 control) (1 μg/ml; Alexis Biochemicals, San Diego, CA). Cell viability was evaluated 48 h after the addition of the cytotoxic agent to the media by measuring lactate dehydrogenase (LDH) activity in the culture-conditioned media. LDH activity was measured spectrophotometrically in the culture media, following nicotinamide adenine dinucleotide (reduced form) oxidation at 340 nm. Total LDH activity was defined as the sum of intracellular and extracellular LDH activity. Cytotoxicity was evaluated as percentage of total LDH activity. Each experiment was performed in triplicate and repeated at least twice.
Evaluation of Apoptosis
Terminal deoxynucleotidyltransferase-mediated dUTP nick end labelling (TUNEL) was performed using the kit purchased by Roche Molecular Biochemicals according to the manufacturer's instructions. The TUNEL-positive cells were counted in eight different fields for each well taken from experiments that were run in triplicate.
For May-Grunwald–Giemsa's staining, cells were washed in phosphate-buffered saline (PBS) solution, fixed with May-Grunwald's staining containing methanol for 3 min, then washed with a 200 mM NaH2PO4/Na2HPO4 buffer (pH 7.4) for additional 3 min. Cells were then stained (3 min) with Giemsa, and air-dried after three/four rapid washes in water. Cells were then analyzed under fluorescence microscope with × 40 objective and photographed.
Immunofluorescence
Cells were fixed in methanol for 5 min, raised in PBS and then incubated overnight with a polyclonal anti-TRAIL (Alexis) used at 1:500 dilution, a monoclonal anti-p53 antibody (Ab 8, Neomarker) used at 1:300 dilution, or anti-DR5 (Alexis) used at 1:400 dilution. CyTM3-conjugated secondary antibodies were used. Slides were mounted with moviol and were examined using a fluorescence microscope.
Western Blot Analysis
Cells were harvested in 100 μl of lysis buffer containing 50 mM Tris, pH 7.6, 150 mM NaCl, 5 mM ethylene diaminetetraacetic acid, 1 mM phenyl-methyl-sulfonyl-fluoride, 0.5 μg/μl leupeptin, 5 μg/μl aprotinin, 1 μg/ml pepstatin. The samples were sonicated and centrifuged at 15 000g for 30 min at 4°C. The resulting supernatants were isolated and the protein content was determined by a conventional method (BCA protein assay Kit, Pierce, Rockford, IL). Fifteen micrograms of total protein were electrophoresed on 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and transferred to nitrocellulose paper (Schleicher and Schuell, Dassel, Germany). Filters were incubated at room temperature overnight with anti-TRAIL (the same used for immunohistochemistry), anti-p53 (the same used for immunohistochemistry), anti-DR5 (C-20 Santa Cruz Biotechnology), or anti-tubulin (Ab3, Neo Markers) antibody in 3% nonfat dried milk (Sigma). The secondary antibodies (Santa Cruz Biotechnology) and a chemiluminescence blotting substrate kit (Boehringer, Mannheim, Germany) were used for immunodetection. Evaluation of immunoreactivity was performed on immunoblots by densitometric analysis using a KLB 2222–020 Ultra Scan XL laser densitometer at a wavelength of 633 nm.
Statistical Evaluation
Results in cell viability and densitometric analysis of the immunoblots are given as mean±standard error mean values. Statistical significance of differences was determined by mean values of the analysis of variance, following Student's test. Significance was accepted for a p-value of <0.05.
RESULTS
TRAIL Expression is Necessary but Nonsufficient to Mediate Aβ Neurotoxicity
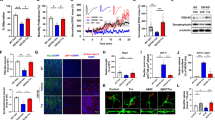
As previously described, high, neurotoxic doses of Aβ25–35 caused an increase expression of TRAIL that was followed by apoptosis (Cantarella et al, 2003). Here, we investigate whether lower doses of Aβ can modulate TRAIL expression on neuronal cell line SH-SY5Y. To that purpose, human SH-SY5Y neuroblastoma cells were differentiated with retinoic acid for 1 week and then challenged with increasing concentrations of Aβ25–35, ranging from 1 nM to 25 μM, to evaluate cell death and TRAIL protein expression. Interestingly, low concentration of Aβ25–35 (10 nM) was able to induce a significant increase of TRAIL expression and this effect proportionally enhanced with the increase of Aβ doses (Figure 1, panels a and b).

Expression of TRAIL after exposure to increasing concentrations of Aβ25–35. Cells were treated with different Aβ concentration and then immunofluorescence analysis with polyclonal anti-TRAIL antibody and cell viability were performed. (a) Representative immunostaining carried out with anti-TRAIL antibody. (b) Quantitative analysis of TRAIL immunofluorescence staining. Values are expressed as integrate optical density (IOD) and represent means±SEM of at least three different experiments from three separate cell preparations. *p<0.01 vs the corresponding control values. (c) May-Grunwald–Giemsa's staining that visualized the late phase of apoptosis following concentration curve of Aβ. Scale bar=50 μm. (d) LDH measurement following concentration curve of Aβ. Data were expressed as percentage of cell viability, and represent means±SEM of at least three different experiments and are from three separate cell preparations. *p<0.01 vs the corresponding control values.
Cell death was characterized by May-Grunwald–Giemsa's staining to visualize the late phase of apoptosis and by measurement of extracellular LDH release. In line with previous observations (Mattson et al, 1992; Weiss et al, 1994), the neurotoxic effects induced by Aβ25–35 were found to be concentration-dependent, being undetectable at 10 nM. Very few scattered apoptotic nuclei with a reduction of cell viability of about 10% were present at 1 μM concentration, whereas the highest dose of 25 μM induced about 40% of cell death with evident sign of apoptosis (Figure 1, panels c and d). Altogether, these data raised the question of why, after exposure of the cells to low concentrations of Aβ, the increased expression of TRAIL was not followed by apoptosis. Because TRAIL toxicity is mediated by its death receptors, we examined the expression of the death receptor DR5 after different Aβ concentrations. Figure 2, panel a shows a representative Western blot analysis performed on neuronal cells, carried out with anti-TRAIL and anti-DR5 antibodies. In line with the immunofluorescence data reported above, TRAIL protein levels increased progressively with the increase of Aβ doses. On the other hand, DR5 expression was virtually absent in untreated cells and as well as in the cells exposed to 10 nM or 1 μM Aβ. The DR5 protein levels significantly increased only with toxic concentration (25 μM) of Aβ peptide (Figure 2, panel a). In this set of experiments, we also examined the expression of the tumor suppressor p53, because it is well recognized to be involved in Aβ neurotoxicity and to be a transcriptional inducer of the DR5 gene (Wu et al, 1997). Like DR5, the p53 protein levels appeared significantly enhanced only at the toxic concentration of Aβ (Figure 2, panel a). The quantitative analysis of immunoblot experiment performed on three different cell culture preparations confirmed the immunofluorescence data showing that Aβ promotes a progressive concentration-dependent increase of TRAIL, but only at the proapoptotic concentration increases of DR5 and p53 expression (Figure 2, panel b). These data suggest a critical role of the death receptor DR5 in TRAIL-mediated Aβ neurotoxicity.

Expression of TRAIL, DR5, and p53 after exposure to increasing concentrations of Aβ25–35. Cells were treated with different Aβ concentrations and then a Western blot analysis was carried out with polyclonal anti-TRAIL, polyclonal anti-DR5, monoclonal anti-p53, or monoclonal anti-β tubulin (β tub) antibodies was performed. (a) Representative immunoblot. (b) Densitometric analysis of immunoblots performed with anti-TRAIL (open bar), anti-DR5 (grey bar), or anti-p53 (black bar) antibodies. Values are expressed as integrated density of respective antibody signal over β tubulin value and represent means±SEM of at least three different experiments from three separate cell preparations. *p<0.01 vs the corresponding control values.
DR5 Expression is Mediated by p53 in Human Neuronal Cell Line and Mouse Cortical Neurons
Neuronal cell lines were preincubated with 200 nM PIF, a compound known to inhibit p53 activity and then exposed to 25 μM Aβ25–35 or to 10 μM Aβ1–42. At 8 h after the addition of Aβ to the medium, the cells were processed for immunofluorescence analysis carried out with anti-DR5 and anti-p53 antibodies.
Both Aβ25–35 and Aβ1–42 induced a significant increase of DR5 immunoreactivity that was prevented by the preincubation with PIF (Figure 3, panel a). Also, p53 immunostaining was found enhanced after Aβ25–35 and Aβ1–42 treatments, and the presence of PIF did not alter Aβ-induced increase of p53 immunoreactivity (Figure 3, panel b). These results are consistent with those of Culmsee et al (2001), who suggest that the mechanism of action of PIF is the inhibition of p53 nuclear translocation and prevention of its DNA-binding activity, rather than the downregulation of newly synthesized p53 or the inhibition of its activation by relevant kinases.

Effects of PIF on DR5 and p53 expression following Aβ injury. Representative immunofluorescence analysis carried out with polyclonal anti-DR5 antibody (a) and monoclonal anti-p53 antibody (b) in neuronal cell lines treated with 25 μM Aβ25–35, or 10 μM Aβ1–42 in the absence or presence of 200 nM PIF. Representative immunofluorescence analysis carried out with polyclonal anti-DR5 antibody (c) and monoclonal anti-p53 antibody (d) in cortical neurons treated with 10 μM Aβ1–42 in the absence or presence of 200 nM PIF. Scale bar=50 μm. (e) Representative Western blot analysis with polyclonal anti-DR5 antibody in cortical neurons treated with 10 μM Aβ1–42 in the absence or presence of 200 nM PIF. (f) Densitometric analysis of DR5 immunoblot performed on cells treated 10 μM Aβ1–42 in the absence (open bar) or presence of 200 μM PIF (black bar). Values are expressed as integrated density of DR5 signal over β tubulin value and represent means±SEM of at least three different experiments from three separate cell preparations. *p<0.01 vs the corresponding control values.
Similar results were obtained with cortical neurons exposed to 10 μM Aβ1–42. An intense cytoplasmic staining was observed in cortical neurons treated with Aβ1–42 at the dose of 10 μM and immunostained with anti-DR5 antibody. Similarly to what was found in neuronal cell line, the inhibition of p53 activity by PIF prevented the induction of DR5 expression induced by Aβ1–42 (Figure 3, panel c). Furthermore, many cortical neurons exposed to Aβ1–42 accumulated p53 protein in the nuclear compartment, whereas in some cells p53 remained confined to the cytoplasm. Pretreatment with PIF did not modify p53 induction induced by Aβ1–42 (Figure 3, panel d). In support to immunofluorescence data, Western blot analysis confirmed the direct correlation between p53 and DR5 receptor. In fact, an intense band representing DR5 protein levels was observed after Aβ1–42 exposure. DR5 protein levels returned to basal levels after the combined treatment with PIF and Aβ1–42. Differently, PIF did not change the increased p53 protein levels induced by Aβ1–42 (Figure 3, panel e). The quantitative analysis performed on three different cell culture preparations confirmed the data observed in the representative Western blot analysis. Both DR5 and p53 protein levels were increased after treatment of the cells with Aβ1–42. The pretreatment with PIF prevented only Aβ1–42-induced increase of DR5 but not p53 expression (Figure 3, panel f).
PIF and Anti-DR5 Blocking Peptide Prevent Aβ Neurotoxicity in Neuronal Cell Line and Cortical Neurons
To give more insight into the intracellular cascades activated by Aβ, neuronal cell lines and cortical neurons were treated with Aβ peptides in the presence or absence of PIF or the anti-DR5 blocking peptide and then they were evaluated for cell viability. Treatment of neuronal cell line with Aβ25–35 25 μM resulted in a reduction of cell viability by about 40%. A dose curve with fibrillar Aβ1–42 was also performed. The fibrillar peptide Aβ1–42 reduced cell viability in a concentration-dependent manner (data not show). At the dose of 10 μM Aβ1–42 peptide cause a 65% of cell death. PIF partially but significantly rescued Aβ25–35 and Aβ1–42-induced cell death (Figure 4, panel a). Interestingly, blockade of DR5 receptor with a specific anti-DR5 antibody completely prevented Aβ-induced cell death. These effects were specific as treatment of the cells with a control isotype IgG1 did not affect Aβ-induced reduction of cell viability (Figure 4, panel b). Similar results were obtained with cortical neurons treated with Aβ1–42. The neurotoxic effects of Aβ1–42 were partially abolished in cortical neurons pretreated with PIF (Figure 5, panel A). Cortical neurons were also treated with Aβ1–42 with or without anti-DR5 antibody and 24 h later a TUNEL analysis was performed. As shown in Figure 5, panel B, few scattered TUNEL-positive cells were observed in untreated cells or in cells treated only with anti-DR5 antibody (panel a and b). TUNEL-positive cells were found very abundant in the sample treated with Aβ1–42 alone (Figure 5, panel Bc), whereas the combined treatment of Aβ with anti-DR5 antibody showed a pattern of cell viability similar to that found in untreated cells with very few apoptotic nuclei (Figure 5, panel Bd). These results were confirmed by LDH release measurement, showing that anti-DR5 antibody prevented the cell loss induced by Aβ1–42 (Figure 5, panel C).

Effects of PIF or the DR5 blocking peptide on cell viability following Aβ injuries in human neuronal cell line. LDH release was measured in the culture media of cells treated with saline (open bars), 25 μM Aβ25–35 (grey bars) or 10 μM Aβ1–42 (dark bars) in the absence or presence of 200 nM PIF (a), or the DR5 blocking peptide (1 μg/ml) (b). IgG1 isotype was used as control. Values represent means±SEM of at least three different experiments and are from three separate cell preparations. *p<0.01 vs the corresponding untreated control values.

Effects of PIF or the DR5 blocking peptide on cell viability after Aβ injuries in primary cortical neurons. LDH release was measured in the culture media of cells treated with saline (open bars) or 10 μM Aβ1–42 (dark bars) in the absence or presence of 200 nM PIF (A), or the DR5 blocking peptide (1 μg/ml) (C). Values represent means±SEM of at least three different experiments and are from three separate cell preparations. (B) TUNEL analysis of cortical neurons untreated (a), treated with the DR5 blocking peptide antibody (1 μg/ml) alone (b), 10 μM Aβ1–42 (c) or 10 μM Aβ1–42 plus the DR5 blocking peptide (1 μg/ml) (d). *p<0.01 vs the corresponding control values.
DR5 Receptor is Expressed in AD Brain
We previously found a significant expression of TRAIL protein in the brain of AD patients, suggesting an involvement of TRAIL death pathway in the neurodegenerative process of AD (Uberti et al, 2004). Different studies have also demonstrated the presence of TRAIL death receptor in human brain (Dorr et al, 2002; Nitsch et al, 2000). We had the opportunity to evaluate the expression of DR5 receptor in the brain of three AD patients. The patients showed a history of AD, as designed according to the Diagnostic and Statistical Manual of Mental Disorders (fourth edition) and the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer's Disease and Related Disorders Association clinical criteria. Postmortem examination confirmed the diagnosis of AD because the presence of abundant Congo red-stained amyloid deposits and neurofibrillary tangles; hematoxylin–eosin staining also demonstrated marked cell loss (data not shown). Some cortical brain slices were also analyzed for DR5 expression with an anti-DR5 antibody. The DR5-like immunoreactivity was then compared with the one found in the brain obtained from an age-matched non-AD subject. As shown in a representative picture of brain slices derived from the sporadic AD patient (Figure 6), consistent and significant DR5 immunoreactivity was found in the AD brain (panel b). At high magnification, the signal was found to be confined on cytoplasmic edge with a punctate staining (panel d). Similar results were obtained on brain slices derived from the two familiar case of AD (data not shown). In brain slices from the control subjects, DR5 immunoreactivity was indeed found although at very low levels (panels a and c).

Expression of DR5 receptor in AD human brain cortex. Representative images of cerebral cortex from one non-AD (a, c) and one AD (b, d) subjects immunostained with polyclonal anti-DR5 antibody. Panels c and d represent high magnification of non-AD and AD cortical slices, respectively. Arrows indicate DR5 immunoreactivity. Scale bar=500 μm.
DISCUSSION
It is well recognized that AD is a multifactorial disease in which many events occurring either separately or sequentially can contribute to the neurodegenerative process. In this scenario, Aβ seems to play a pivotal role. However, up to now, the mechanism behind Aβ-induced neurodegeneration is still controversial (Yankner et al, 1989; Geula et al, 1998). The difficulty in understanding the Aβ effects is sustained also by the fact that this toxic peptide can activate simultaneously different pathways that proceed separately or converge together, and ultimately all lead to neuronal death. Identification of a preferential pathway could definitely be useful to design a new therapeutic drug to slow the progression of AD pathology.
Here, we show that the exposure of differentiated SH-SY5Y neuronal cells or primary cortical neurons to Aβ peptide results in the induction of TRAIL, its death receptor DR5, and p53. A dose–response study of the changes in TRAIL, p53, and DR5 expression demonstrated that Aβ displays concentration-related effects characterized by the sequential involvement of the three factors. In particular, low, nontoxic concentrations of Aβ (nanomolar range) induced the expression of TRAIL without changing DR5 and p53 protein levels. High, neurotoxic concentrations of Aβ (micromolar range) caused an increased expression of all three proteins examined. Thus, in our experimental model, TRAIL was a necessary but not sufficient factor to induce cell death upon Aβ exposure.
In this model, DR5 receptor appears to play a critical role as its expression was found to increase only after exposure to the highest, neurotoxic concentration of Aβ. It has been shown that TRAIL binds to five specific receptors belonging to the TNF/NGF family (Pan et al, 1997a; Walczak et al, 1997). However, only two of them, DR4 and DR5, have an intracellular death domain, which appears essential for triggering cell death processes (Pan et al, 1997a; Walczak et al, 1997). In mice only DR5 receptor has been identified (Wu et al, 1999). This receptor is a homolog of human DR5 and DR4 and binds TRAIL with an affinity similar to the one displayed by human DR4 and DR5 receptors. In this study, DR4 was not taken into consideration because we previously demonstrated that in differentiated SH-SY5Y cells, Aβ25–35 as well as TRAIL synthetic peptide did not alter the DR4 expression (Cantarella et al, 2003). The most convincing result indicating the crucial role of DR5 in Aβ neurotoxicity comes from the experiments showing that the DR5 blocking peptide completely prevents Aβ-induced neurotoxicity in both neuronal cell line and cortical neurons.
The earlier biochemical event following the engagement of DRs receptors by their ligand is the recruitment of protein towards the intracellular death domain of the receptor to form a structure known as the death-inducing signalling complex (DISC) (Kischkel et al, 1995). DISC includes the recruitment of FADD and the activation of pro-caspase 8 or pro-caspase 10. Caspase 8 activated the pro-caspase 3 and in turn they lead to apoptosis. Furthermore, caspase 8 can induce the cleavage of bid, which translocates to the mitochondria where it activates bax and bak, providing a mechanism for crosstalk between the DRs and the apoptotic intrinsic pathway.
The correlation between DR5 and p53 comes from the identification of a subtractive hybridization screen for p53-target genes indicating DR5 gene as one of them. We found that the increased expression of DR5 after Aβ was prevented by PIF, a compound that inhibited p53 activity. p53 is a pleiotropic transcription factor that plays an important role in determining cell fate under certain conditions (see review Almog and Rotter, 1997). p53 is constitutively present in many cell types including neurons and is upregulated and activated via phosphorylation following various insults including excitotoxicity, ischemic injury, ionizing radiation, and oxidative stress (Crumrine et al, 1994; Sakhi et al, 1997; Uberti et al, 1998; Johnson et al, 1998; Inamura et al, 2000). Aβ-induced cytotoxicity is also demonstrated to involve p53 in primary cortical neurons (Copani et al, 2001) and different neuronal cell lines (Misiti et al, 2005; Matsumoto et al, 2006). In line with these latter studies, our data obtained with PIF demonstrated that Aβ neurotoxicity is mediated by p53 in both neuronal cell line and cortical neurons. In fact, inhibition of p53 activation prevented the Aβ-induced neurotoxicity. PIF is a synthetic inhibitor of p53 and was shown to protect against radiation, excitotoxicity, and, as here demonstrated, against Aβ toxicity (Culmsee et al, 2001). The postulated mechanism of PIF activity involved the inhibition of p53 nuclear translocation and prevention of its binding to specific DNA sites and it is not related to downregulation of newly synthesized p53 or inhibition of p53 activation by relevant kinases (Culmsee et al, 2001). In line with these evidences, we have shown that PIF did not alter Aβ-induced increase of p53 expression whereas it prevented the increased expression of DR5.
p53 promotes cellular apoptosis through transactivation of various pro-apoptotic genes that can act via an intrinsic pathway involving the mitochondria activation or an extrinsic pathway requiring activation of death receptors. Although the extrinsic pathway and the intrinsic pathway for apoptosis are capable of operating independently, accumulated evidence suggests that a crosstalk between the two pathways exists in cells (for a review see Green, 1998). The results obtained with anti-DR5 antibody indicated that the blocking of DR5 receptor completely prevented Aβ-induced neurotoxicity in neuronal cultures. These data suggest that p53-mediated apoptosis may occur by a preferential activation of death receptors pathway. We cannot exclude the involvement of intrinsic apoptotic pathway in Aβ neurotoxicity. Indeed, our data suggest that the death receptor pathway, through the activation of caspase 8, can also activate the intrinsic pathway through the cleavage of bid. The truncated Bid then translocates to mitochondria and triggers cytochrome c release (Li et al, 1998; Luo et al, 1998). It has been proposed that Bid regulates cytochrome c release by inducing Bax translocation into mitochondria. In this regard, cells lacking Bax were resistant to TRAIL-induced apoptosis (Deng and Wu, 2000).
Finally, this study demonstrated a significant expression of DR5 receptor in AD brain. The expression of apoptosis-mediating TRAIL receptors indicates that brain cells, including neurons, are potentially susceptible to TRAIL. Although it is unknown whether or not other TRAIL receptors are modulated in AD pathology, the upregulation of TRAIL death receptor DR5 together with the presence of TRAIL in AD brain (Uberti et al, 2004) generate an appropriate environment to execute a cell death program. A possible role of TRAIL pathway in neurodegenerative diseases was also suggested by Martin-Villalba et al (1999) who showed that, after middle cerebral artery occlusion in adult rats, TRAIL is expressed in the apoptotic areas of the postischemic brain. Furthermore, TRAIL was found to be one of the major apoptotic factors inducing neuronal death in a murine model of HIV central nervous system infection (Miura et al, 2003; Ryan et al, 2004).
In conclusion, these data suggest a role of TRAIL death pathway in Aβ neurotoxicity and propose DR5 receptor as a possible target for the development of neuroprotective drugs in those neuropathologies in which TRAIL pathway plays a relevant role.
References
Almog N, Rotter V (1997). Involvement of p53 in cell differentiation and development. Biochim Biophys Acta 1333: F1–F27.
Cantarella G, Uberti D, Carsana T, Lombardo G, Bernardini R, Memo M (2003). Neutralization of TRAIL death pathway protects human neuronal cell line from beta-amyloid toxicity. Cell Death Differ 10: 134–141.
Copani A, Uberti D, Sortino MA, Bruno V, Nicoletti F, Memo M (2001). Activation of cell-cycle-associated proteins in neuronal death: a mandatory or dispensable path? Trends Neurosci 24: 25–31.
Cotman CW, Pike CJ, Copani A (1992). Beta amyloid neurotoxicity: a discussion of in vitro findings. Neurobiol Aging 13: 587–590.
Crumrine RC, Thomas AL, Morgan PF (1994). Attenuation of p53 expression protects against focal ischemic damage in transgenic mice. J Cereb Blood Flow Metab 14: 887–891.
Culmsee C, Zhu X, Yu QS, Chan SL, Camandola S, Guo Z et al (2001). A synthetic inhibitor of p53 protects neurons against death induced by ischemic and excitotoxic insults, and amyloid beta-peptide. J Neurochem 77: 220–228.
Degli-Esposti MA, Dougall WC, Smolak PJ, Waugh JY, Smith CA, Goodwin RG (1997). The novel receptor TRAIL-R4 induces NF-kappaB and protects against TRAIL-mediated apoptosis, yet retains an incomplete death domain. Immunity 7: 813–820.
Deng Y, Wu X (2000). Peg3/Pw1 promotes p53 mediated apoptosis by inducing Bax translocation from cytosol to mitochondria. Proc Natl Acad Sci 97: 12050–12055.
Dewachter I, Van Dorpe J, Smeijers L, Gilis M, Kuiperi C, Laenen I et al (2000). Aging increased amyloid peptide and caused amyloid plaques in brain of old APP/V717I transgenic mice by a different mechanism than mutant presenilin1. J Neurosci 20: 6452–6458.
Dorr J, Bechmann I, Waiczies S, Aktas O, Walczak H, Krammer PH et al (2002). Lack of tumor necrosis factor-related apoptosis-inducing ligand but presence of its receptors in the human brain. J Neurosci 22: 1–5.
Ekinci FJ, Linsley M, Shea TB (2000). Beta amyloid-induced calcium influx induces apoptosis in culture by oxidative stress rather than tau phosphorylation. Mol Brain Res 76: 389–395.
Emery JG, McDonnell P, Burke MB, Deen KC, Lyn S, Silverman C et al (1998). Osteoprotegerin is a receptor for the cytotoxic ligand TRAIL. J Biol Chem 273: 14363–14367.
Geula C, Wu CK, Saroff D, Lorenzo A, Yuan M, Yankner BA (1998). Aging renders the brain vulnerable to amyloid beta-protein neurotoxicity. Nat Med 4: 827–831.
Gray CW, Patel AJ (1995). Neurodegeneration mediated by glutamate and beta-amyloid peptide: a comparison and possible interaction. Brain Res 691: 169–179.
Green DR (1998). Apoptotic pathway: the road to ruin. Cell 94: 695–698.
Haas C, Scholossmacher MC, Hung AY, Vigo-Pelfrey C, Mellon A, Ostaszewski BL et al (1992). Amyloid beta-peptide is produced by cultured cells during normal metabolism. Nature 359: 149–153.
Helpern JA, Lee SP, Falangola MF, Dyakin VV, Bogart A, Ardekani B et al (2004). MRI assessment of neuropathology in a transgenic mouse model of Alzheimer's disease. Magn Reson Med 51: 794–798.
Heppner FL, Gandy S, McLaurin J (2004). Current concepts and future prospects for Alzheimer disease vaccines. Alzheimer Dis Assoc Disord 18: 38–43.
Ichikawa K, Liu W, Zhao L, Wang Z, Liu D, Ohtsuka T et al (2001). Tumoricidal activity of a novel anti-human DR5 monoclonal antibody without hepatocyte cytotoxicity. Nat Med 7: 954–960.
Inamura N, Araki T, Enokido Y, Nishio C, Aizawa S, Hatanaka H (2000). Role of p53 in DNA strand break-induced apoptosis in organotypic slice culture from the mouse cerebellum. J Neurosci Res 60: 450–457.
Johnson MD, Xiang H, London S, Kinoshita Y, Knudson M, Mayberg M et al (1998). Evidence for involvement of Bax and p53, but not caspases, in radiation-induced cell death of cultured postnatal hippocampal neurons. J Neurosci Res 54: 721–733.
Kischkel FC, Hellbardt S, Behrmann I, Germer M, Pawlita M, Krammer PH et al (1995). Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J 14: 5579–5588.
Kowalska A (2003). Amyloid precursor protein gene mutations responsible for early-onset autosomal dominant Alzheimer's disease. Folia Neuropathol 41: 35–40.
LeVine III H (2004). The Amyloid Hypothesis and the clearance and degradation of Alzheimer's beta-peptide. J Alzheimers Dis 6: 303–314.
Lewis HD, Beher D, Smith D, Hewson L, Cookson N, Reynolds DS et al (2004). Novel aspects of accumulation dynamics and A beta composition in transgenic models of AD. Neurobiol Aging 21: 1175–1185.
Li P, Nijhawan D, Budihardjo I, Sirinvasula C, Wang X (1998). Cytochrome c and dATP-dependent formation of Apaf-1/caspase 9 complex initiates an apoptotic protease caspase. Cell 91: 479–489.
Luo X, Budihardjo I, Zou H, Slaughter C, Wang X (1998). BID, a Blc2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 94: 481–490.
Martin-Villalba A, Herr I, Jeremias I, Hahne M, Brandt R, Vogel J et al (1999). CD95 (FAS-L/APO-1L) and Tumour Necrosis Factor-Related Apoptosis-Inducing Ligand mediate ischemia-induced apoptosis in neurons. J Neurosci 15: 3809–3817.
Matsumoto K, Akao Y, Yi H, Shamoto-Nagai M, Maruyama W, Naoi M (2006). Overexpression of amyloid precursor protein induces susceptibility to oxidative stress in human neuroblastoma SH-SY5Y cells. J Neural Transm 113: 125–135.
Mattson MP, Cheng B, Davis D, Bryant K, Lieberburg I, Rydel RE (1992). beta-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J Neurosci 12: 376–389.
Milani D, Zauli G, Rimondi E, Celeghini C, Marmiroli S, Narducci P et al (2003). Tumour necrosis factor-related apoptosis-inducing ligand sequentially activates pro-survival and pro-apoptotic pathways in SK-N-MC neuronal cells. J Neurochem 86: 126–135.
Mirra SS, Heyman A, McKell D, Sumi SM, Crain BJ, Brownlee LM et al (1991). The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathological assessment of Alzheimer's disease. Neurology 41: 479–486.
Misiti F, Sampaolese B, Pezzotti M, Marini S, Coletta M, Ceccarelli L et al (2005). Abeta(31–35) peptide induce apoptosis in PC 12 cells: contrast with Abeta(25–35) peptide and examination of underlying mechanisms. Neurochem Int 46: 575–583.
Miura Y, Misawa N, Kawano Y, Okada H, Inagaki Y, Yamamoto N et al (2003). Tumour necrosis factor-related apoptosis-inducing ligand induces neuronal death in a murine model of HIV central nervous system infection. Proc Natl Acad Sci USA 100: 2777–2782.
Moechars D, Lorent K, Van Leuven F (1999). Premature death in transgenic mice that overexpress a mutant amyloid precursor protein is preceded by severe neurodegeneration and apoptosis. Neuroscience 91: 819–830.
Mori H, Takio K, Ogawara M, Selkoe DJ (1992). Mass spectometry of purified amyloid beta protein in Alzheimer's disease. J Biol Chem 267: 17082–17086.
Nitsch R, Bechmann I, Deisz RA, Haas D, Lehmann TN, Wendling U et al (2000). Human brain-cell death induced by tumour-necrosis-factor-related apoptosis-inducing ligand (TRAIL). Lancet 356: 827–828.
Oster-Granite ML, McPhie DL, Greenan J, Neve RL (1996). Age-dependent neuronal and synaptic degeneration in mice transgenic for the C terminus of the amyloid precursor protein. J Neurosci 16: 6732–6741.
Pan G, Ni J, Wei Y, Yu G, Gentz R, Dixit VM (1997b). An antagonist decoy receptor and a death domain-containing receptor for TRAIL. Science 277: 815–818.
Pan G, O'Rourke K, Chinnaiyan AM, Gentz R, Ebner R, Ni J et al (1997a). The receptor for the cytotoxic ligand TRAIL. Science 276: 111–113.
Peng QL, Buz'Zard AR, Lau BHS (2002). Pycnogenol protects neurons from amyloid beta peptide-induced apoptosis. Mol Brain Res 104: 55–65.
Pike CJ, Burdik D, Walencewicz AJ, Gable CG, Cotman CW (1993). Neurodegeneration induced by β-amyloid peptides in vitro: the role of peptide assembly state. J Neurosci 13: 1676–1687.
Pitti RM, Marsters SA, Ruppert S, Donahue CJ, Moore A, Ashkenazi A (1996). Induction of apoptosis by Apo-2 ligand, a new member of the tumor necrosis factor cytokine family. J Biol Chem 271: 12687–12690.
Rosales-Corral S, Tan DX, Reiter RJ, Valdivia-Velazquez M, Acosta-Martinez JP, Ortiz GG (2004). Kinetics of the neuroinflammation-oxidative stress correlation in rat brain following the injection of fibrillar amyloid beta onto the hippocampus in vivo. J Neuroimmunol 150: 20–28.
Ryan LA, Peng H, Erichsen DA, Huang Y, Persidsky Y, Zhou Y et al (2004). TNF-related apoptosis-inducing ligand mediates human neuronal apoptosis: link to HIV-1-associated dementia. J Neuroimmunol 148: 127–139.
Sakhi S, Bruce A, Sun N, Tocco G, Baudry M, Schreiber SS (1997). Induction of tumor suppressor p53 and DNA fragmentation in organotypic hippocampal cultures following excitotoxin treatment. Exp Neurol 145: 81–88.
Selkoe DJ (1996). Amyloid beta protein and the genetics of Alzheimer's disease. J Biol Chem 271: 18295–18298.
Seubert PC, Vigo-Pelfrey C, Esch F, Lee M, Dovery H, Davis D et al (1992). Isolation and quantification of soluble Alzheimer's beta-peptide from biological fluids. Nature 359: 325–327.
Sheridan JP, Marsters S, Pitti RM, Gurney A, Skubatch M, Baldwin D et al (1997). Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors. Science 277: 818–821.
Tamagno E, Parola M, Gulglielmotto M, Santoro G, Bardini P, Marra L et al (2003). Multiple signaling events in amyloid beta induced oxidative stress-dependent neuronal apoptosis. Free Radical Biol Med 35: 45–58.
Uberti D, Belloni M, Grilli M, Spano P, Memo M (1998). Induction of tumour-suppressor phosphoprotein p53 in the apoptosis of cultured rat cerebellar neurones triggered by excitatory amino acids. Eur J Neurosci 10: 246–254.
Uberti D, Cantarella G, Facchetti F, Cafici A, Grasso G, Bernardini R et al (2004). TRAIL is expressed in the brain cells of Alzheimer's disease patients. Neuroreport 15: 579–581.
Walczak H, Degli-Esposti MA, Johnson RS, Smolak PJ, Waugh JY, Boiani N et al (1997). TRAIL-R2: a novel apoptosis-mediating receptor for TRAIL. EMBO J 16: 5386–5397.
Weiss JH, Pike CJ, Cotman CW (1994). Ca2+ channel blockers attenuate beta-amyloid peptide toxicity to cortical neurons in culture. J Neurochem 62: 372–375.
Wu GS, Burns TF, McDonald III ER, Jiang W, Meng R, Krantz ID et al (1997). KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene. Nat Genet 17: 141–143.
Wu GS, Burns TF, Zhan Y, Alnemri ES, El-Deiry S (1999). Molecular cloning and functional analysis of the mouse homologue of the KILLER/DR5 tumour necrosis factor-related apoptosis –inducing ligand (TRAIL) death receptor. Cancer Res 59: 2770–2775.
Yankner BA, Dawes LR, Fisher S, Villa-Komaroff L, Oster-Granite ML, Neve RL (1989). Neurotoxicity of a fragment of the amyloid precursor associated with Alzheimer's disease. Science 245: 417–420.
Zatti G, Gidoni R, Barbiero L, Binetti G, Pozzan T, Fasolato C et al (2004). The presinilin 2 M239I mutation associated with familial Alzheimer's disease reduces Ca2+ release from intracellular stores. Neurobiol Dis 15: 269–278.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Uberti, D., Ferrari-Toninelli, G., Bonini, S. et al. Blockade of the Tumor Necrosis Factor-Related Apoptosis Inducing Ligand Death Receptor DR5 Prevents β-Amyloid Neurotoxicity. Neuropsychopharmacol 32, 872–880 (2007). https://doi.org/10.1038/sj.npp.1301185
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.npp.1301185
Keywords
This article is cited by
-
The immune system on the TRAIL of Alzheimer’s disease
Journal of Neuroinflammation (2020)
-
Beneficial effects of curtailing immune susceptibility in an Alzheimer’s disease model
Journal of Neuroinflammation (2019)
-
Clinical perspectives of TRAIL: insights into central nervous system disorders
Cellular and Molecular Life Sciences (2016)
-
TRAIL death receptors DR4 and DR5 mediate cerebral microvascular endothelial cell apoptosis induced by oligomeric Alzheimer’s Aβ
Cell Death & Disease (2012)
-
Neutralization of Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand Reduces Spinal Cord Injury Damage in Mice
Neuropsychopharmacology (2010)
