
Abstract
Stimulants such as methylphenidate and amphetamine are currently the most common treatment for attention deficit hyperactivity disorder (ADHD). For years, it was assumed that stimulants had paradoxical calming effects in ADHD patients, whereas stimulating ‘normal’ individuals and producing locomotor activation in rats. It is now known that low doses of stimulants focus attention and improve executive function in both normal and ADHD subjects. Furthermore, the seminal work of Kuczenski and Segal showed that low, oral doses of methylphenidate reduce locomotor activity in rats as well. Berridge et al have now shown that these low doses produce marked increases in norepinephrine and dopamine release in the prefrontal cortex, whereas having only subtle effects on subcortical catecholamine release. ihe prefrontal cortex regulates behavior and attention using representational knowledge, and imaging and neuropsychological studies have shown that the prefrontal cortex is weaker in subjects with ADHD. This cortical area is very sensitive to levels of catecholamines: moderate levels engage postsynaptic α2A-adrenoceptors and D1 receptors and improve prefrontal regulation of behavior and attention, while high levels impair prefrontal function via α1-adrenoceptors and excessive D1 receptor stimulation. Administering low doses of methylphenidate to rats improves the working memory and attentional functions of the prefrontal cortex, while high doses impair working memory and produce a perseverative pattern of errors similar to that seen in patients. The low dose improvement is hiocked by either an α2-adrenoceptor or Dl receptor antagonist, suggesting that both norepinephrine and dopamine contribute to the beneficial actions of stimulant medications.
Similar content being viewed by others

INTRODUCTION
For decades, basic research on the neurochemical and behavioral actions of stimulants has focused on higher doses that induce locomotor hyperactivity and promote dopamine release in ventral and/or dorsal striatum. This body of work has been the foundation for many of the dopaminergic hypotheses of schizophrenia and drug abuse. In particular, the sensitizing effects of repeated, high-dose stimulant administration have been a major focus of this work. Much of David Segal's career was spent characterizing the behavioral and neurochemical differences that emerge following repeated, high-dose amphetamine administration (see, for example, Segal and Kuczenski, 1987, 1997a, 1997b)
In contrast to these basic studies in rats, humans given stimulant medications for the treatment of attention deficit hyperactivity disorder (ADHD) display reduced locomotor activity and improved attentional focus. For years, it was presumed that stimulant medications had paradoxical effects in ADHD. However, it is now established that the focusing effects of stimulants in ADHD are not paradoxical; these agents have the same effect in ‘normal’ human subjects (albeit a more subtle response given ceiling effects) (Rapoport and Inoff-Germain, 2002). However, the discrepancies between locomotor-activating effects in rodents and the focusing effects in humans remained: it was assumed that the locomotor-activating effects of stimulants in rodents must be owing to species differences, thus weakening the validity of rodent models of ADHD.
A major advance for the field of ADHD was the finding by Kuczenski and Segal (2002) that low, oral doses of methylphenidate—doses that produce blood levels similar to those in ADHD patients—can decrease locomotor activity in juvenile rats. Thus, it was not that rats and humans had opposite responses to stimulants, but rather that rats had previously been given doses far in excess of those equivalent to human therapeutic medications. This key finding redirected the entire field, and has allowed us to study the therapeutic mechanisms of stimulant medications using valid rodent models.
THE BIOCHEMICAL EFFECTS OF LOW-DOSE STIMULANT ADMINISTRATION
Previous research with higher doses of stimulants focused on the dopaminergic effects of these compounds in subcortical structures. Methylphenidate and amphetamine were known to block the dopamine transporter and enhance dopamine release, and robust increases in dopamine release were observed in the nucleus accumbens (Segal and Kuczenski, 1999) and dorsal striatum (Kuczenski and Segal, 1997) of rats given high doses of stimulants. Supportive evidence was found in human imaging studies, where intravenous stimulant administration displaced D2 receptor PET ligands, an indication of increased endogenous dopamine release (Volkow et al, 2002b). This measure of D2 receptor stimulation correlated with measures of reinforcement, consistent with the rodent work (Volkow et al, 2002b). In contrast, oral administration of lower doses of stimulants produces more subtle and slower effects on striatal dopamine release (Volkow et al, 2002a). The amount of dopamine release in the nucleus accumbens is especially pertinent to drug abuse, and thus rodent studies focused on this brain region to try to determine whether the doses of stimulants given to children would alter dopamine release in this structure. Kuczenski and Segal (2002, 2005) first identified the dose regimen of orally administered, low doses that produced blood levels similar to those observed in children taking stimulants to treat ADHD symptoms. They found that these low, oral doses had little or no effect on DA release in the nucleus accumbens, and they found no evidence of stimulant sensitization following low-dose chronic usage. These results in rats are consistent with observations of children taking stimulants: ADHD medications do not produce euphoria (indeed, dysphoria is the more likely side effect), and the incidence of drug abuse is actually reduced in properly medicated ADHD patients (Hechtman and Greenfield, 2003; Katusic et al, 2005).
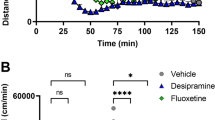
Although Kuczenski and Segal (2002) found only subtle effects of low-dose oral methylphenidate on dopamine release in the nucleus accumbens, they did observe increased release of norepinephrine in the hippocampus. Based on this seminal paper, Berridge et al (2006) then explored low-dose stimulant effects on dopamine and norepinephrine release in the prefrontal cortex, a brain region closely linked to ADHD, as well as in the nucleus accumbens and medial septal area. A summary of these results can be seen in Figure 1. As with Kuczenski and Segal, only subtle effects were observed in accumbens dopamine release. There was also some subcortical norepinephrine release in the medial septal area. However, the greatest effects were observed in prefrontal cortex, where there were especially high levels of norepinephrine release (400% increase; Figure 1a), and significant levels of dopamine release (250% increase; Figure 1b). The functional ramifications of enhanced catecholamine release in prefrontal cortex are discussed below. It is noteworthy that atomoxetine (Strattera) also increases norepinephrine and dopamine release in the prefrontal cortex (Bymaster et al, 2002); thus, catecholamine release in the prefrontal cortex may be a common action for many ADHD therapeutics. It should be noted that available PET and SPECT neuroreceptor ligands are generally unable to detect the delicate catecholamine input to cortex. Thus, current imaging methods are limited to the striatum. Research is in progress to visualize catecholamine actions in the prefrontal cortex of human subjects.

Effects of low-dose methylphenidate (MPH) on extracellular levels of NE and DA within and outside the PFC. (a) NE levels within the PFC and the medial septal area (MSA). (b) DA levels within the PFC and nucleus accumbens (ACC). In each panel, the mean (±SEM) NE or DA levels within 16-min samples collected before (negative numbers) and following (positive numbers) injection of vehicle or 0.5 mg/kg (i.p.) MPH are displayed. Values are expressed as a percentage of baseline (calculated as the average of four pre-infusion samples). Low-dose MPH produces substantially larger increases in NE and DA within the PFC relative to the MSA and Acc, respectively. At this dose, but not 0.25 mg/kg, MPH produces a larger increase in PFC NE levels relative to PFC DA. Similar preferential actions of MPH on PFC catecholamines were observed with oral administration (2.0 mg/kg). *P<0.05 compared to vehicle-treated animals. Data from Berridge et al (2006).
THE ROLE OF THE PREFRONTAL CORTEX IN EXECUTIVE FUNCTIONS
The prefrontal cortex guides behavior and attention using working memory, applying representational knowledge to inhibit inappropriate actions, thoughts, and feelings. These processes are the basis of the so-called executive functions, including regulation of attention, planning, impulse control, mental flexibility, and the initiation and monitoring of action. Lesions to the prefrontal cortex produce symptoms such as forgetfulness, distractibility, impulsivity, perseveration, and disorganization. Lesions in the ventromedial prefrontal cortex can impair regulation of emotion, and result in inappropriate social behaviors such as aggression (Anderson et al, 1999).
The role of prefrontal cortex in attention regulation has been appreciated for many years. The prefrontal cortex regulates what we attend to based on represented goals. The prefrontal cortex inhibits responses to distracting stimuli and suppresses irrelevant thoughts such as proactive interference. Thus, patients with prefrontal cortex lesions are easily distracted (Woods and Knight, 1986; Godefroy and Rousseaux, 1996), are impaired at gating sensory stimuli (Knight et al, 1989; Yamaguchi and Knight, 1990), have poor concentration and organization, and are more vulnerable to disruption from proactive interference (Thompson-Schill et al, 2002). Prefrontal cortex lesions impair the ability to sustain attention, particularly over long delays (Wilkins et al, 1987). Lesions of the dorsolateral prefrontal cortex impair the ability to shift attentional set (Manes et al, 2002). Prefrontal cortex lesions also impair divided attention, and these attentional deficits have been associated with lesions in the left, superior prefrontal cortex (Godefroy and Rousseaux, 1996). Similar results have been seen in animals, where prefrontal cortex lesions impair attentional regulation in monkeys (Malmo, 1942; Bartus and Levere, 1977; Dias et al, 1996), and rats (Muir et al, 1996).
The prefrontal cortex also allows us to regulate overt behaviors including locomotor activity. The right inferior prefrontal cortex is particularly important for behavioral inhibition (reviewed in Aron et al, 2004). Both imaging (Konishi et al, 1999; Rubia et al, 2003) and lesion studies indicate that the right prefrontal cortex in humans is critical for inhibitory abilities, for example, performance of the stop or go–no go tasks. The importance of the prefrontal cortex to inhibitory control has also been shown in monkeys with lesions to the prefrontal cortex (Petrides, 1986), and in electrophysiological (Watanabe, 1986) and imaging studies (Morita et al, 2004). There is also a classic literature demonstrating that prefrontal cortex lesions cause locomotor hyperactivity in monkeys (Kennard et al, 1941; French, 1959; Gross, 1963; Gross and Weiskrantz, 1964). Thus, some of the locomotor hyperactivity observed in ADHD may arise from prefrontal cortex dysfunction.
The cellular basis for prefrontal cortical regulation of attention and behavior has been an arena of intensive research in animals. Prefrontal neurons are able to represent spatial and feature information in the absence of environmental stimulation (Goldman-Rakic, 1995). This firing is often measured during the delay period of working memory tasks when information must be kept in mind for correct performance. Prefrontal neurons can also fire in relationship to an abstract rule that is used to govern action (Wallis et al, 2001). A unique feature of prefrontal neurons is their ability to maintain information in the presence of interference from distracting stimuli (Miller et al, 1993). Delay-related firing also can serve as the basis for behavioral inhibition, as examined in an anti-saccade task in which monkeys must look away from a remembered visual stimulus (Funahashi et al, 1993). Thus, delay-related activity is observed both when an animal must make a memory-guided action and when an animal must withhold a prepotent response based on representational knowledge. Weakening of these abilities likely contributes to ADHD.
PREFRONTAL CORTICAL DYSFUNCTION IN ADHD
Evidence from a variety of perspectives demonstrates that prefrontal cortical function is weaker in subjects with ADHD (reviewed in Arnsten et al, 1996; Barkley, 1997; Aron and Poldrack, 2005). Neuropsychological analyses have shown that patients with ADHD are impaired on the same tasks as those with prefrontal lesions, for example, tasks of behavioral inhibition, reward reversal, and working memory (Itami and Uno, 2002; Bedard et al, 2003; McLean et al, 2004). Although some neuropsychological studies disagree with the importance of executive function deficits in ADHD, these studies are often flawed by the use of inappropriate tasks for evaluating children (eg the Stroop color-naming interference task, which assumes that reading is a prepotent response that must be inhibited, an assumption often invalid in children; van Mourik et al, 2005), disagreement on which processes constitute executive functions (eg not including attention regulation parameters as executive functions; Schoechlin and Engel, 2005), or by the use of tasks with ceiling effects for this patient population. Deficits in attention can also lead to secondary impairments on widespread cognitive abilities that depend on proper perception and encoding. Thus, it is difficult to ascertain specificity in this patient population (Boonstra et al, 2005). It is likely that ADHD does not involve global prefrontal deficits in all ADHD patients, as subcircuits within prefrontal cortex are likely differentially sensitive to genetic mutations and/or developmental abnormalities. This heterogeneity in types of PFC dysfunction would account for a weaker effect size on any one task. However, when viewed as a whole, the literature indicates that functions dependent on the PFC are weaker in ADHD patients. Numerous structural imaging studies have shown reduced size of the prefrontal cortex in ADHD patients, particularly in the right hemisphere (Castellanos et al, 1996; Casey et al, 1997; Filipek et al, 1997; Giedd et al, 2001; Kates et al, 2002; Hill et al, 2003; Sowell et al, 2003). Many of these studies have also found reduced volume of cerebellum, caudate, and corpus callosum (Seidman et al, 2005; Castellanos et al, 2002). A large study of children aged 5–18 showed that the developmental trajectories for these structures, except caudate, remained parallel for patients and controls during childhood and adolescence, and were observed in children who had never received stimulant treatment (Castellanos et al, 2002). These findings emphasize the biological nature of ADHD. Functional imaging studies have also shown evidence of inefficient or reduced blood flow or metabolism in prefrontal cortex of ADHD patients, deficits that correspond with poor prefrontal cortical cognitive function (Rubia et al, 1999; Yeo et al, 2000; Bush et al, 2005).
GENETICS OF ADHD: FOCUS ON CATECHOLAMINES
ADHD is often an inherited disorder, and many of the genes associated with ADHD code for molecules involved with catecholamine transmission (reviewed in Faraone et al, 2005). Initial genetic studies focused on molecules involved with dopamine transmission. Researchers have consistently found associations between ADHD and the genes encoding for the dopamine transporter, and the D1, D4, and D5 receptors. More recently, genetic studies have turned to genes associated with norepinephrine transmission. The norepinephrine synthetic enzyme dopamine beta hydroxylase (DBH) and the gene encoding for the α2A adrenoceptor have been associated with ADHD (Roman et al, 2003; Park et al, 2005). It should be noted that the D4 receptor has very high affinity for norepinephrine (Van Tol et al, 1991) and should really be considered a catecholamine receptor. Patients with ADHD show a greater prevalence of the seven repeat polymorphism form of the D4 receptor that renders this receptor less effective. Intriguingly, a recent study has associated changes in the D4 receptor with reduced prefrontal volume (Durston et al, 2005), and with poor performance of a sustained attention task (Bellgrove et al, 2005). Similarly, genetic alterations in DBH are associated with impaired sustained attention in ADHD (Bellgrove et al, 2006). These relationships between genetic changes in catecholamine signaling and prefrontal cognitive deficits are consistent with the important role of dopamine and norepinephrine in prefrontal cognitive function.
CATECHOLAMINE MODULATION OF PREFRONTAL CORTICAL FUNCTION
The working memory abilities of the prefrontal cortex are greatly influenced by the levels of catecholamines released in prefrontal cortex. Either insufficient or excessive catecholamine receptor stimulation can markedly impair prefrontal cortex cognitive function. For more extensive reviews of this topic, see Arnsten and Robbins (2002) and Arnsten and Li (2005).
Dopamine
Dopamine modulates prefrontal cortex functions through actions at the D1 (D1, D5) and D2 (D2, D3, D4) families of receptors. Research elucidating the role of the D2 family is still in progress, but a clear picture is emerging with regard to the D1 receptor family. There are no drugs that distinguish between D1 and D5 receptors, and thus all references to ‘D1’ generally refer to either the D1 or D5 subtype. Stimulation of D1/D5 receptors produces an inverted ‘U’-shaped dose–response on the working memory and attention regulation processes of the prefrontal cortex (Zahrt et al, 1997; Granon et al, 2000). Whereas modest levels of D1 receptor stimulation are essential for prefrontal cortex function, high levels of dopamine release occur during exposure to stress, and impair working memory and attention regulation. Many of these D1/D5 actions appear to be mediated via cAMP/protein kinase A intracellular signaling mechanisms (Arnsten et al, 2005). A similar inverted ‘U’ has been described at the cellular level in monkeys performing a spatial working memory task. Moderate levels of D1/D5 receptor stimulation suppress delay-related firing for nonpreferred spatial directions (ie ‘noise’), and thus enhance spatial tuning (Williams and Goldman-Rakic, 1995; Vijayraghavan et al, 2006). However, high levels of D1/D5 receptor stimulation suppress delay-related firing for all directions, and thus erode spatial tuning (Vijayraghavan et al, 2006). Limited studies have been performed in humans owing to the lack of selective D1/D5 compounds available for human use. However, studies with nonselective compounds have suggested that an inverted ‘U’ may be evident in humans as well as animals (Kimberg et al, 1997), and that compounds that prefer D1 receptors may be more helpful than D2 agonists in improving working memory (Muller et al, 1998). Taken together, these studies suggest that genetic alterations in the D1 or D5 receptor, or in molecules that influence the level of DA available in the synapse (eg the dopamine transporter, the catabolic enzyme COMT) could influence the strength of prefrontal cortical regulation of behavior.
Norepinephrine
As with dopamine, moderate levels of NE are critical for proper prefrontal cortical function, whereas high levels released during stress impair prefrontal cortical function. The beneficial effects of NE may be especially relevant to medications used to treat ADHD (see below). Norepinephrine improves prefrontal cortex function through actions at post-synaptic, α2A receptors. Although the older literature emphasized the prevalence of pre-synaptic α2 receptors that inhibit NE release and reduce noradrenergic cell firing, it is now known that the vast majority of α2 receptors are localized post-synaptic to norepinephrine terminals. It is these post-synaptic receptors that mediate the enhancing effects of α2 agonists on prefrontal cognitive function (Arnsten and Goldman-Rakic, 1985; Cai et al, 1993). Studies in genetically modified mice have demonstrated that the α2A subtype, rather than the α2B or α2C subtype, underlies the enhancement in prefrontal cognitive function (Franowicz et al, 2002). These enhancing effects appear to result from inhibiion of cAMP intracellular signaling (Ramos et al, 2006). The α2A agonist, guanfacine, improves many aspects of prefrontal cortical function, including working memory, attention regulation, behavioral inhibition, and planning, and these effects have been seen in rats (Tanila et al, 1996; Ramos et al, 2006), monkeys (Arnsten et al, 1988; Mao et al, 1999), and humans (Jakala et al, 1999). The enhancing effects of α2A agonists are most evident in subjects with PFC dysfunction, and these compounds can be less efficacious and/or potent in young adult animals (Franowicz and Arnsten, 1998) or humans (Muller et al, 2005) with healthy endogenous noradrenergic systems. SPECT imaging has shown that systemic administration of guanfacine activates dorsolateral prefrontal cortex in monkeys performing a spatial working memory task (Mao et al, 1999; Avery et al, 2000). Importantly, guanfacine appears to improve the functioning of both lateral (Mao et al, 1999; Wang et al, 2004) and ventromedial (Steere and Arnsten, 1997) prefrontal circuits, and thus strengthens regulation of both intellectual and emotional responses. α2A-Adrenoceptor stimulation also strengthens prefrontal function at the cellular level. Thus, α2A agonists enhance, whereas antagonists erode, delay-related prefrontal cell firing (Sawaguchi, 1998; Li et al, 1999; Wang et al, 2006). In contrast to stimulation of DA D1 receptors that suppress ‘noise’, norepinephrine α2A receptor stimulation enhances spatial tuning by increasing ‘signals’, that is, increasing delay-related firing for the preferred spatial direction. Studies in progress indicate that guanfacine increases delay-related firing and improves prefrontal cognitive function by strengthening the functional connectivity of microcircuits in the prefrontal cortex via inhibition of cAMP intracellular signaling (Wang et al, 2006). In contrast, blockade of α2 receptors in the prefrontal cortex of monkeys erodes delay-related cell firing and recreates all the symptoms of ADHD: poor impulse control (Ma et al, 2003), locomotor hyperactivity (Ma et al, 2005), and weakened working memory (Li and Mei, 1994) underlying increased distractibility. From these basic studies, it is clear that genetic alterations in NE synthetic enzymes (DBH) or in α2A adrenoceptors may contribute to ADHD symptomology by weakening endogenous noradrenergic α2A-adrenoceptor signaling.
In contrast to these beneficial effects at moderate concentrations, high levels of norepinephrine release (eg during stress) impair prefrontal function through actions at α1 receptors (Birnbaum et al, 1999, 2004) and possibly β1 receptors as well (Ramos et al, 2005). Based on this work, the α1 receptor blocker, prazosin, is now in use for the treatment of post-traumatic stress disorder (Raskind et al, 2003). Recordings of prefrontal neurons have found that α1-adrenoceptor stimulation decreases delay-related cell firing for the preferred direction (Birnbaum et al, 2004). These detrimental actions are mediated by activation of protein kinase C intracellular signaling (Birnbaum et al, 2004). Interestingly, excessive protein kinase C signaling has been associated with mania (Manji and Lenox, 1999), and bipolar disorder and ADHD are often confused in children with high levels of distractibility, impulsivity, and hyperactivity (Biederman et al, 2000). In this regard, it is of interest that most ‘atypical’ anti-psychotic medications have potent α1 blocking properties. Thus, prefrontal deficits associated with high levels of stress or with bipolar disorder may benefit from inhibition of α1/protein kinase C signaling.
LOW-DOSE STIMULANTS IMPROVE PREFRONTAL CORTICAL FUNCTION IN HUMANS AND ANIMALS
Given the role of prefrontal cortical deficits in ADHD, studies have examined whether stimulant medication can ameliorate prefrontal deficits in patients and in animals. Methylphenidate has now been found to improve spatial working memory, response inhibition, set-shifting, and other prefrontal cognitive functions in both ‘normal’ college students (Elliott et al, 1997; Mehta et al, 2000) and in children and adults with ADHD (Aron et al, 2003; Bedard et al, 2003; Mehta et al, 2004). In adults with ADHD, childhood ratings of ADHD correlated with response to methylphenidate on the spatial working memory task (Turner et al, 2005). Thus, studies of spatial working memory performance are likely relevant to the therapeutic actions of this compound in ADHD.
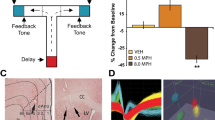
Spatial working memory is classically assessed in rats using delayed alternation testing on a T maze (Larsen and Divac, 1978). Low doses of methylphenidate, which increase catecholamine release in prefrontal cortex, have recently been shown to improve delayed alternation performance in rats (Figure 2; Arnsten and Dudley, 2005; Berridge et al, 2006). Low doses of methylphenidate also improved performance of a sustained attention task, the five-choice attention task that similarly depends upon prefrontal cortex (Berridge et al, 2006). In contrast to low doses of methylphenidate, higher doses impaired performance of the delayed alternation task (eg Figure 2a), consistent with excessive catecholamine receptor stimulation impairing prefrontal cortical cognitive function. These impairing effects at higher doses may correspond to the mental inflexibility often described in patients taking excessive doses of stimulant medication.

The effects of oral administration of MPH on delayed alternation performance in male rats. (a) A representative dose–response curve from an individual rat. For most rats, a lower dose (1.0–2.0 mg/kg, p.o. 30 min) was found to improve performance, whereas higher doses often impaired performance (1.5–3.0 mg/kg). (b) The enhancing effects of methylphenidate (1.0–2.0 mg/kg) were blocked by co-administration of the α2 adrenoceptor antagonist idazoxan (IDA, 0.1 mg/kg), which had no effect on its own. Results represent mean±SEM percent correct on the delayed alternation task. **significantly different from VEH; †significantly different from MPH. (c) The enhancing effects of methylphenidate were blocked by co-administration of the dopamine D1 receptor antagonist SCH23390 at doses (0.01–0.1 mg/kg) that had no effect on their own. **Significantly different from VEH; †significantly different from MPH. Data adapted from Arnsten and Dudley (2005).
The receptor mechanisms underlying the therapeutic effects of low, oral doses of methylphenidate have recently begun to be examined. As shown in Figure 2, studies in rats have shown that the cognitive-enhancing effects of methylphenidate on spatial working memory are prevented by blockade of either noradrenergic α2-adrenoceptors (Figure 2b) or dopamine D1 receptors (Figure 2c) (Arnsten and Dudley, 2005). Thus, stimulants likely facilitate endogenous catecholamine stimulation of D1 and α2A receptors in prefrontal cortex. The receptor mechanisms underlying the cognitive errors observed following high-dose methylphenidate administration are currently under investigation. These may involve excessive D1, α1, and/or β1 receptor stimulation in prefrontal cortex. Thus, we now have rodent models to examine the neurochemistry and neuropharmacology of stimulant actions in ADHD. The results so far, when viewed in combination with genetic studies of ADHD, suggest that stimulants may ameliorate weaknesses in catecholamine signaling in the prefrontal cortex of ADHD subjects.
SUMMARY
Thanks to the patient work of Kuczenski and Segal (2002), the correct path has been illumined to pursue the neural mechanisms underlying the therapeutic effects of stimulant medications in rodent models. The spotlight has turned to much lower doses, and a logical picture emerges, bridging genetics and neurobiology with the pharmacological treatment of ADHD.
A final comment—those who knew David observed his extraordinary energy, focus, and commitment to his work and family. I would like to thank him for teaching me to be a dedicated scientist and a devoted parent, all at once.
References
Anderson SW, Bechara A, Damasio H, Tranel D, Damasio AR (1999). Impairment of social and moral behavior related to early damage in human prefrontal cortex. Nat Neurosci 2: 1032–1037.
Arnsten AFT, Cai JX, Goldman-Rakic PS (1988). The alpha-2 adrenergic agonist guanfacine improves memory in aged monkeys without sedative or hypotensive side effects. J Neurosci 8: 4287–4298.
Arnsten AFT, Dudley AG (2005). Methylphenidate improves prefrontal cortical cognitive function through a2 adrenoceptor and dopamine D1 receptor actions: relevance to therapeutic effects in Attention Deficit Hyperactivity Disorder. Behav Brain Funct (Biomed Central) 1: 2.
Arnsten AFT, Goldman-Rakic PS (1985). Alpha-2 adrenergic mechanisms in prefrontal cortex associated with cognitive decline in aged nonhuman primates. Science 230: 1273–1276.
Arnsten AFT, Li B-M (2005). Neurobiology of executive functions: catecholamine influences on prefrontal cortical function. Biol Psychiatry 57: 1377–1384.
Arnsten AFT, Ramos B, Birnbaum SB, Taylor JR (2005). Protein kinase A as a therapeutic target for memory disorders: rationale and challenges. Trends Mol Med 11: 121–128.
Arnsten AFT, Robbins TW (2002). Neurochemical modulation of prefrontal cortical function in humans and animals. In: Stuss DT, Knight RT (eds). Principles of Frontal Lobe Function. Oxford University Press: New York. pp 51–84.
Arnsten AFT, Steere JC, Hunt RD (1996). The contribution of alpha-2 noradrenergic mechanisms to prefrontal cortical cognitive function: potential significance to Attention Deficit Hyperactivity Disorder. Arch Gen Psychiatry 53: 448–455.
Aron AR, Dowson JH, Sahakian BJ, Robbins TW (2003). Methylphenidate improves response inhibition in adults with attention-deficit/hyperactivity disorder. Biol Psychiatry 54: 1465–1468.
Aron AR, Poldrack RA (2005). The cognitive neuroscience of response inhibition: relevance for genetic research in attention-deficit/hyperactivity disorder. Biol Psychiat 57: 1285–1292.
Aron AR, Robbins TW, Poldrack RA (2004). Inhibition and the right inferior frontal cortex. Trends Cogn Sci 8: 170–177.
Avery RA, Franowicz JS, Studholme C, van Dyck CH, Arnsten AFT (2000). The alpha-2A-adenoceptor agonist, guanfacine, increases regional cerebral blood flow in dorsolateral prefrontal cortex of monkeys performing a spatial working memory task. Neuropsychopharmacology 23: 240–249.
Barkley RA (1997). ADHD and the Nature of Self-Control. Guilford Press: New York.
Bartus RT, Levere TE (1977). Frontal decortication in rhesus monkeys: a test of the interference hypothesis. Brain Res 119: 233–248.
Bedard AC, Ickowicz A, Logan GD, Hogg-Johnson S, Schachar R, Tannock R (2003). Selective inhibition in children with attention-deficit hyperactivity disorder off and on stimulant medication. J Abnorm Child Psychol 31: 315–327.
Bellgrove MA, Hawi Z, Gill M, Robertson IH (2006). The cognitive genetics of attention deficit hyperactivity disorder (ADHD): sustained attention as a candidate phenotype. Cortex (in press).
Bellgrove MA, Hawi Z, Lowe N, Kirley A, Robertson IH, Gill M (2005). DRD4 gene variants and sustained attention in attention deficit hyperactivity disorder (ADHD): effects of associated alleles at the VNTR and -521 SNP. Am J Med Genet B Neuropsychiat Genet 136: 81–86.
Berridge CW, Devilbiss DM, Andrzejewski ME, Arnsten AFT, Kelley AE, Schmeichel B et al (2006). Methylphenidate preferentially increases catecholamine neurotransmission within the prefrontal cortex at low doses that enhance cognitive function. Biol Psychiatry [Epub ahead of print].
Biederman J, Mick E, Faraone SV, Spencer T, Wilens TE, Wozniak J (2000). Pediatric mania: a developmental subtype of bipolar disorder? Biol Psychiatry 48: 458–466.
Birnbaum SB, Yuan P, Bloom A, Davis D, Gobeske K, Sweatt D et al (2004). Protein kinase C overactivity impairs prefrontal cortical regulation of working memory. Science 306: 882–884.
Birnbaum SG, Gobeske KT, Auerbach J, Taylor JR, Arnsten AFT (1999). A role for norepinephrine in stress-induced cognitive deficits: alpha-1-adrenoceptor mediation in prefrontal cortex. Biol Psychiatry 46: 1266–1274.
Boonstra AM, Oosterlaan J, Sergeant JA, Buitelaar JK (2005). Executive functioning in adult ADHD: a meta-analytic review. Psychol Med 35: 1097–1108.
Bush G, Valera EM, Seidman LJ (2005). Functional neuroimaging of attention-deficit/hyperactivity disorder: a review and suggested future directions. Biol Psychiatry 57: 1273–1284.
Bymaster FP, Katner JS, Nelson DL, Hemrick-Luecke SK, Threlkeld PG, Heiligenstein JH et al (2002). Atomoxetine increases extracellular levels of norepinephrine and dopamine in prefrontal cortex of rat: a potential mechanism for efficacy in attention deficit/hyperactivity disorder. Neuropsychopharmacology 27: 699–711.
Cai JX, Ma Y, Xu L, Hu X (1993). Reserpine impairs spatial working memory performance in monkeys: reversal by the alpha-2 adrenergic agonist clonidine. Brain Res 614: 191–196.
Casey BJ, Castellanos FX, Giedd JN, Marsh WL, Hamburger SD, Schubert AB et al (1997). Implication of right frontostriatal circuitry in response inhibition and attention-deficit/hyperactivity disorder. J Am Acad Child Adolescent Psychiatry 36: 374–383.
Castellanos FX, Giedd JN, Marsh WL, Hamburger SD, Vaituzis AC, Dickstein DP et al (1996). Quantitative brain magnetic resonance imaging in attention deficit/hyperactivity disorder. Arch Gen Psychiatry 53: 607–616.
Castellanos FX, Lee PP, Sharp W, Jeffries NO, Greenstein DK, Clasen LS et al (2002). Developmental trajectories of brain volume abnormalities in children and adolescents with attention-deficit/hyperactivity disorder. JAMA 288: 1740–1748.
Dias R, Roberts A, Robbins TW (1996). Dissociation in prefrontal cortex of affective and attentional shifts. Nature 380: 69–72.
Durston S, Fossella JA, Casey BJ, Hulshoff Pol HE, Galvan A, Schnack HG et al (2005). Differential effects of DRD4 and DAT1 genotype on fronto-striatal gray matter volumes in a sample of subjects with attention deficit hyperactivity disorder, their unaffected siblings, and controls. Mol Psychiatry 10: 678–685.
Elliott R, Sahakian BJ, Matthews K, Bannerjea A, Rimmer J, Robbins TW (1997). Effects of methylphenidate on spatial working memory and planning in healthy young adults. Psychopharmacology 131: 196–206.
Faraone SV, Perlis RH, Doyle AE, Smoller JW, Goralnick JJ, Holmgren MA et al (2005). Molecular genetics of attention-deficit/hyperactivity disorder. Biol Psychiatry 57: 1313–1323.
Filipek PA, Semrud-Clikeman M, Steingard RJ, Renshaw PF, Kennedy DN, Biederman J (1997). Volumetric MRI analysis comparing subjects having attention-deficit hyperactivity disorder with normal controls. Neurology 48: 589–601.
Franowicz JCS, Arnsten AFT (1998). The alpha-2A noradrenergic agonist, guanfacine, improves delayed response performance in young adult rhesus monkeys. Psychopharmacology 136: 8–14.
Franowicz JS, Kessler L, Dailey-Borja CM, Kobilka BK, Limbird LE, Arnsten AFT (2002). Mutation of the alpha2A-adrenoceptor impairs working memory performance and annuls cognitive enhancement by guanfacine. J Neurosci 22: 8771–8777.
French GM (1959). Locomotor effects of regional ablation of frontal cortex in rhesus monkeys. J Comp Physiol Psychol 52: 18–24.
Funahashi S, Chafee MV, Goldman-Rakic PS (1993). Prefrontal neuronal activity in rhesus monkeys performing a delayed anti-saccade task. Nature 365: 753–756.
Giedd JN, Blumenthal J, Molloy E, Castellanos FX (2001). Brain imaging of attention deficit/hyperactivity disorder. Ann NY Acad Sci 931: 33–49.
Godefroy O, Rousseaux M (1996). Divided and focused attention in patients with lesion of the prefrontal cortex. Brain Cognition 30: 155–174.
Goldman-Rakic PS (1995). Cellular basis of working memory. Neuron 14: 477–485.
Granon S, Passetti F, Thomas KL, Dalley JW, Everitt BJ, Robbins TW (2000). Enhanced and impaired attentional performance after infusion of D1 dopaminergic receptor agents into rat prefrontal cortex. J Neurosci 20: 1208–1215.
Gross CG (1963). Locomotor activity following lateral frontal lesions in rhesus monkeys. J Comp Physiol Psychol 56: 232–236.
Gross CG, Weiskrantz L (1964). Some changes in behavior produced by lateral frontal lesions in the macaque. In: Warren JM, Akert K (eds). The Frontal Granular Cortex and Behavior. McGraw-Hill Book Co: New York. pp 74–101.
Hechtman L, Greenfield B (2003). Long-term use of stimulants in children with attention deficit hyperactivity disorder: safety, efficacy, and long-term outcome. Paediatr Drugs 5: 787–794.
Hill DE, Yeo RA, Campbell RA, Hart B, Vigil J, Brooks W (2003). Magnetic resonance imaging correlates of attention-deficit/hyperactivity disorder in children. Neuropsychology 17: 496–506.
Itami S, Uno H (2002). Orbitofrontal cortex dysfunction in attention-deficit hyperactivity disorder revealed by reversal and extinction tasks. NeuroReport 13: 2453–2457.
Jakala P, Riekkinen M, Sirvio J, Koivisto E, Kejonen K, Vanhanen M et al (1999). Guanfacine, but not clonidine, improves planning and working memory performance in humans. Neuropsychopharmacology 20: 460–470.
Kates WR, Frederikse M, Mostofsky SH, Folley BS, Cooper K, Mazur-Hopkins P et al (2002). MRI parcellation of the frontal lobe in boys with attention deficit hyperactivity disorder or Tourette syndrome. Psychiatry Res 116: 63–81.
Katusic SK, Barbaresi WJ, Colligan RC, Weaver AL, Leibson CL, Jacobsen SJ (2005). Psychostimulant treatment and risk for substance abuse among young adults with a history of attention-deficit/hyperactivity disorder: a population-based, birth cohort study. J Child Adolesc Psychopharmacol 15: 764–776.
Kennard MA, Spencer S, Fountain G (1941). Hyperactivity in monkeys following lesions of the frontal lobes. J Neurophysiol 4: 512–524.
Kimberg DY, D'Esposito M, Farah MJ (1997). Effects of bromocriptine on human subjects depend on working memory capacity. NeuroReport 8: 3581–3585.
Knight RT, Scabini D, Woods DL (1989). Prefrontal cortex gating of auditory transmission in humans. Brain Res 504: 338–342.
Konishi S, Nakajima K, Uchida I, Kikyo H, Kameyama M, Miyashita Y (1999). Common inhibitory mechanism in human inferior prefrontal cortex revealed by event-related functional MRI. Brain 122: 981–991.
Kuczenski R, Segal DS (1997). Effects of methylphenidate on extracellular dopamine, serotonin, and norepinephrine: comparison with amphetamine. J Neurochem 68: 2032–2037.
Kuczenski R, Segal DS (2002). Exposure of adolescent rats to oral methylphenidate: preferential effects on extracellular norepinephrine and absence of sensitization and cross-sensitization to methamphetamine. J Neurosci 22: 7264–7271.
Kuczenski R, Segal DS (2005). Stimulant actions in rodents: implications for attention-deficit/hyperactivity disorder treatment and potential substance abuse. Biol Psychiatry 57: 1391–1396.
Larsen JK, Divac I (1978). Selective ablations within the prefrontal cortex of the rat and performance of delayed alternation. Physiol Psychol 6: 15–17.
Li B-M, Mao Z-M, Wang M, Mei Z-T (1999). Alpha-2 adrenergic modulation of prefrontal cortical neuronal activity related to spatial working memory in monkeys. Neuropsychopharmacology 21: 601–610.
Li B-M, Mei Z-T (1994). Delayed response deficit induced by local injection of the alpha-2 adrenergic antagonist yohimbine into the dorsolateral prefrontal cortex in young adult monkeys. Behav Neural Biol 62: 134–139.
Ma C-L, Arnsten AFT, Li B-M (2005). Locomotor hyperactivity induced by blockade of prefrontal cortical alpha-2-adrenoceptors in monkeys. Biol Psychiatry 57: 192–195.
Ma C-L, Qi X-L, Peng J-Y, Li B-M (2003). Selective deficit in no-go performance induced by blockade of prefrontal cortical alpha2-adrenoceptors in monkeys. NeuroReport 14: 1013–1016.
Malmo RB (1942). Interference factors in delayed response in monkeys after removal of frontal lobes. Neurophysics 5: 295–308.
Manes F, Sahakian BJ, Clark L, Rogers R, Antoun N, Aitken M et al (2002). Decision-making processes following damage to the prefrontal cortex. Brain 125: 624–639.
Manji HK, Lenox RH (1999). Protein kinase C signaling in the brain: molecular transduction of mood stabilization in the treatment of manic-depressive illness. Biol Psychiatry 46: 1328–1351.
Mao Z-M, Arnsten AFT, Li B-M (1999). Local infusion of alpha-1 adrenergic agonist into the prefrontal cortex impairs spatial working memory performance in monkeys. Biol Psychiatry 46: 1259–1265.
McLean A, Dowson J, Toone B, Young S, Bazanis E, Robbins TW et al (2004). Characteristic neurocognitive profile associated with adult attention-deficit/hyperactivity disorder. Psychol Med 34: 681–692.
Mehta MA, Goodyer IM, Sahakian BJ (2004). Methylphenidate improves working memory and set-shifting in AD/HD: relationships to baseline memory capacity. J Child Psychol Psychiatry 45: 293–305.
Mehta MA, Owen AM, Sahakian BJ, Mavaddat N, Pickard JD, Robbins TW (2000). Methylphenidate enhances working memory by modulating discrete frontal and parietal lobe regions in the human brain. J Neurosci 20: RC651–RC656.
Miller EK, Li L, Desimone R (1993). Activity of neurons in anterior inferior temporal cortex during a short-term memory task. J Neurosci 13: 1460–1478.
Morita M, Nakahara K, Hayashi T (2004). A rapid presentation event-related functional magnetic resonance imaging study of response inhibition in macaque monkeys. Neurosci Lett 356: 203–206.
Muir JL, Everitt BJ, Robbins TW (1996). The cerebral cortex of the rat and visual attentional function: dissociable effects of mediofrontal, cingulate, anterior dorsolateral, and parietal cortex lesions on a five-choice serial reaction time task. Cerebral Cortex 6: 470–481.
Muller U, Clark L, Lam ML, Moore RM, Murphy CL, Richmond NK et al (2005). Lack of effects of guanfacine on executive and memory functions in healthy male volunteers. Psychopharmacology 182: 205–213.
Muller U, von Cramon DY, Pollmann S (1998). D1- versus D2-receptor modulation of visuospatial working memory in humans. J Neurosci 18: 2720–2728.
Park L, Nigg JT, Waldman ID, Nummy KA, Huang-Pollock C, Rappley M et al (2005). Association and linkage of alpha-2A adrenergic receptor gene polymorphisms with childhood ADHD. Mol Psychiatry 10: 572–580.
Petrides M (1986). The effect of periarcuate lesions in the monkey on the performance of symmetrically and asymmetrically reinforced visual and auditory go, no-go tasks. J Neurosci 6: 2054–2063.
Ramos B, Colgan L, Nou E, Ovadia S, Wilson SR, Arnsten AFT (2005). The beta-1 adrenergic antagonist, betaxolol, improves working memory performance in rats and monkeys. Biol Psychiatry 58: 894–900.
Ramos B, Stark D, Verduzco L, Arnsten AFT (2006). Guanfacine improves prefrontal cortical reulation of behavior through inhibition of cAMP/PKA signaling. (in submission).
Rapoport JL, Inoff-Germain G (2002). Responses to methylphenidate in attention-deficit/hyperactivity disorder and normal children: update 2002. J Atten Disord 6: S57–S60.
Raskind MA, Peskind ER, Kanter ED, Petrie EC, Radant A, Thompson C et al (2003). Prazosin reduces nightmares and other PTSD symptoms in combat veterans: a placebo-controlled study. Am J Psychiatry 160: 371–373.
Roman T, Schmitz M, Polanczyk GV, Eizirik M, Rohde LA, Hutz MH (2003). Is the alpha-2A adrenergic receptor gene (ADRA2A) associated with attention-deficit/hyperactivity disorder? Am J Med Genet B Neuropsychiat Genet 120: 116–120.
Rubia K, Overmeyer S, Taylor E, Brammer M, Williams SCR, Simmons A et al (1999). Hypofrontality in attention deficit hyperactivity disorder during higher-order motor control: a study with functional MRI. Am J Psychiatry 156: 891–896.
Rubia K, Smith AB, Brammer MJ, Taylor E (2003). Right inferior prefrontal cortex mediates response inhibition while mesial prefrontal cortex is responsible for error detection. Neuroimage 20: 351–358.
Sawaguchi T (1998). Attenuation of delay-period activity of monkey prefrontal cortical neurons by an alpha-2 adrenergic antagonist during an oculomotor delayed-response task. J Neurophysiol 80: 2200–2205.
Schoechlin C, Engel RR (2005). Neuropsychological performance in adult attention-deficit hyperactivity disorder: meta-analysis of empirical data. Arch Clin Neuropsychol 20: 727–744.
Segal DS, Kuczenski R (1987). Individual differences in responsiveness to single and repeated amphetamine administration: behavioral characteristics and neurochemical correlates. J Pharmacol Exp Ther 242: 917–926.
Segal DS, Kuczenski R (1997a). Repeated binge exposures to amphetamine and methamphetamine: behavioral and neurochemical characterization. J Pharmacol Exp Ther 282: 561–573.
Segal DS, Kuczenski R (1997b). An escalating dose ‘binge’ model of amphetamine psychosis: behavioral and neurochemical characteristics. J Neurosci 17: 2551–2566.
Segal DS, Kuczenski R (1999). Escalating dose-binge treatment with methylphenidate: role of serotonin in the emergent behavioral profile. J Pharmacol Exp Ther 291: 19–30.
Seidman LJ, Valera EM, Makris N (2005). Structural brain imaging of attention-deficit/hyperactivity disorder. Biol Psychiatry 57: 1263–1272.
Sowell ER, Thompson PM, Welcome SE, Henkenius AL, Toga AW, Peterson BS (2003). Cortical abnormalities in children and adolescents with attention-deficit hyperactivity disorder. Lancet 362: 1699–1707.
Steere JC, Arnsten AFT (1997). The alpha-2A noradrenergic agonist, guanfacine, improves visual object discrimination reversal performance in rhesus monkeys. Behav Neurosci 111: 1–9.
Tanila H, Rama P, Carlson S (1996). The effects of prefrontal intracortical microinjections of an alpha-2 agonist, alpha-2 antagonist and lidocaine on the delayed alternation performance of aged rats. Brain Res Bull 40: 117–119.
Thompson-Schill SL, Jonides J, Marshuetz C, Smith EE, D'Esposito M, Kan IP et al (2002). Effects of frontal lobe damage on interference effects in working memory. Cogn Affect Behav Neurosci 2: 109–120.
Turner DC, Blackwell AD, Dowson JH, McLean A, Sahakian BJ (2005). Neurocognitive effects of methylphenidate in adult attention-deficit/hyperactivity disorder. Psychopharmacology 178: 286–295.
van Mourik R, Oosterlaan J, Sergeant JA (2005). The Stroop revisited: a meta-analysis of interference control in AD/HD. J Child Psychol Psychiatry 46: 150–165.
Van Tol HHM, Bunzow JR, Guan H-C, Sunahara RK, Seeman P, Niznik HB et al (1991). Cloning of the gene for a human dopamine D4 receptor with high affinity for the antipsychotic clozapine. Nature 350: 610–614.
Vijayraghavan S, Wang M, Birnbaum SG, Bruce CJ, Williams GV, Arnsten AFT (2006). Inverted-U dopamine D1 receptor actions on prefrontal neurons engaged in working memory. Nat Neurosci (in submission).
Volkow ND, Fowler JS, Wang GJ, Ding YS, Gatley SJ (2002a). Role of dopamine in the therapeutic and reinforcing effects of methylphenidate in humans: results from imaging studies. Eur Neuropsychopharmacol 12: 557–566.
Volkow ND, Wang GJ, Fowler JS, Thanos PP, Logan J, Gatley SJ et al (2002b). Brain DA D2 receptors predict reinforcing effects of stimulants in humans: replication study. Synapse 46: 79–82.
Wallis JD, Anderson KC, Miller EK (2001). Single neurons in prefrontal cortex encode abstract rules. Nature 411: 953–956.
Wang M, Ramos B, Paspalas C, Duque A, Vijayraghavan S, Brennan A et al (2006). 2A-Adrenoceptor stimulation enhances working memory by closing Ih channels and strengthening functional connectivity of prefrontal cortical microcircuitry (in preparation).
Wang M, Tang ZX, Li BM (2004). Enhanced visuomotor associative learning following stimulation of alpha 2A-adrenoceptors in the ventral prefrontal cortex in monkeys. Brain Res 1024: 176–182.
Watanabe M (1986). Prefrontal unit activity during delayed conditional go/no-go discrimination in the monkey I. Relation to the stimulus. Brain Res Rev 382: 1–14.
Wilkins AJ, Shallice T, McCarthy R (1987). Frontal lesions and sustained attention. Neuropsychologia 25: 359–365.
Williams GV, Goldman-Rakic PS (1995). Blockade of dopamine D1 receptors enhances memory fields of prefrontal neurons in primate cerebral cortex. Nature 376: 572–575.
Woods DL, Knight RT (1986). Electrophysiological evidence of increased distractability after dorsolateral prefrontal lesions. Neurology 36: 212–216.
Yamaguchi S, Knight RT (1990). Gating of somatosensory input by human prefrontal cortex. Brain Res 521: 281–288.
Yeo RA, Hill D, Campbell R, Vigil J, Brooks WM (2000). Developmental instability and working memory ability in children: a magnetic resonance spectroscopy investigation. Dev Neuropsychol 17: 143–159.
Zahrt J, Taylor JR, Mathew RG, Arnsten AFT (1997). Supranormal stimulation of dopamine D1 receptors in the rodent prefrontal cortex impairs spatial working memory performance. J Neurosci 17: 8528–8535.
Acknowledgements
Some of the research reviewed in this article was funded by R37 AG06036, R21 MH066393, and a research grant from Shire Pharmaceuticals. AFTA and Yale University have a license agreement with Shire Pharmaceuticals for the development of guanfacine for the treatment of ADHD.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Arnsten, A. Stimulants: Therapeutic Actions in ADHD. Neuropsychopharmacol 31, 2376–2383 (2006). https://doi.org/10.1038/sj.npp.1301164
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.npp.1301164
Keywords
This article is cited by
-
Meta-model of Human Recognition-behavioral Adaptation System
Integrative Psychological and Behavioral Science (2024)
-
Male-specific, replicable and functional roles of genetic variants and cerebral gray matter volumes in ADHD: a gene-wide association study across KTN1 and a region-wide functional validation across brain
Child and Adolescent Psychiatry and Mental Health (2023)
-
Adenosine A2A Receptor Blockade Ameliorates Mania Like Symptoms in Rats: Signaling to PKC-α and Akt/GSK-3β/β-Catenin
Molecular Neurobiology (2022)
-
The Role of the Dopamine Transporter in the Effects of Amphetamine on Sleep and Sleep Architecture in Drosophila
Neurochemical Research (2022)
-
Childhood urbanicity interacts with polygenic risk for depression to affect stress-related medial prefrontal function
Translational Psychiatry (2021)
