
Abstract
Repeated moderate doses of methamphetamine (mAMPH) damage forebrain monoaminergic terminals and nonmonoaminergic cells in somatosensory cortex, and impair performance in a novelty preference task of object recognition (OR). This study aimed to determine whether the memory deficit seen after a neurotoxic mAMPH regimen results from damage to dopamine (DA) and/or serotonin (5-HT) terminals. Animals were given a neurotoxic regimen of mAMPH, p-chloroamphetamine (PCA, preferentially damages 5-HT terminals), d-amphetamine (d-AMPH, preferentially damages DA terminals), or saline. After 1 week, animals were trained and tested for OR memory. Rats treated with mAMPH showed no recognition memory during the short-term memory (STM) test, whereas both PCA- and d-AMPH-treated rats showed OR STM scores comparable to controls. After behavioral testing, the specificity of monoaminergic lesions was determined by postmortem [125I]RTI-55 binding to dopamine (DAT) and serotonin (SERT) transporter proteins. Tissue from a separate group of animals killed 3 days after drug treatment was processed for Fluoro-Jade (F-J) fluorescence histochemistry to detect damaged cortical neurons. mAMPH-treated rats showed reductions in striatal DAT and hippocampal (HC) and perirhinal (pRh) SERT, as well as degeneration of neurons in primary somatosensory cortex. In PCA-treated rats, HC and pRh SERT were substantially depleted, but striatal DAT and cortical neuron survival were unaffected. By contrast, d-AMPH-treated animals showed marked depletions in striatal DAT and cortical neurodegeneration, but HC and pRh SERT were unaffected. This pattern of results indicates that no single feature of mAMPH-induced neurotoxicity is sufficient to produce the OR impairments seen after mAMPH treatment.
Similar content being viewed by others

INTRODUCTION
Repeated exposure to moderate-to-high doses of methamphetamine (mAMPH) produces long-term damage to presynaptic dopaminergic and serotonergic terminals in rodents (Wagner et al, 1980; Ricaurte et al, 1980) and in humans (Wilson et al, 1996; McCann et al, 1998). Additionally, mAMPH produces degeneration of nonmonoaminergic cell bodies in somatosensory cortex (Commins and Seiden, 1986; Eisch et al, 1998; O'Dell and Marshall, 2000). The long-term damage to striatal dopaminergic terminals resulting from such mAMPH exposure is well characterized, with the terminal loss greatest in ventral caudate putamen (Eisch et al, 1992). While the cellular and neurochemical repercussions of these mAMPH dosing regimens have been investigated extensively, less is known concerning the cognitive function of humans and animals following repeated mAMPH administrations.
Cognitive impairments have been reported for current (Simon et al, 2002) and detoxified (Volkow et al, 2001a, 2001b) mAMPH users. Additionally, two separate laboratories have reported that rats are impaired in a novelty preference task of object recognition (OR; Ennaceur and Delacour, 1988) following a regimen of mAMPH, which produces decreases in markers of monoaminergic terminal integrity (Bisagno et al, 2002; Schröder et al, 2003). However, it is not clear whether the memory impairment seen after mAMPH-induced neurotoxicity is due to damage to the dopaminergic, or serotonergic systems, both, or neither. The objectives of the present study were (1) to replicate the observation that animals given neurotoxic doses of mAMPH show impairments in OR and (2) to investigate whether OR memory depends on the integrity of the dopaminergic and/or serotonergic system. To this end, neurotoxicity to either dopaminergic or serotonergic terminals was induced using two amphetamine analogues with more specific profiles of damage, p-chloroamphetamine (PCA) and d-amphetamine (d-AMPH). In high doses, d-AMPH damages dopaminergic terminals in the striatum and cell bodies in parietal cortex (Bowyer et al, 1998), while largely sparing serotonin (5-HT) terminals. By contrast, PCA causes long-term depletions in markers of serotonergic activity (Pletscher et al, 1963; Fuller et al, 1965; Sanders-Bush et al, 1972) and structural alterations in serotonergic neurons (Harvey et al, 1975) while largely sparing dopaminergic terminals. At 1 week after completion of the OR task, animals in all three experiments were euthanized and their brains were processed for [125I]RTI-55 binding autoradiography of dopaminergic and serotonergic transporters as indicators of the integrity of forebrain dopaminergic and serotonergic terminals. Additionally, to assess cortical neuronal injury following exposure to the various drug treatments, a subset of the animals in each of the experiments was euthanized and the brains processed for Fluoro-Jade (F-J) fluorescence histochemistry, a technique developed by Schmued et al, (1997); Schmued and Hopkins, (2000) that labels degenerating neurons.
MATERIALS AND METHODS
Subjects
Adult male Sprague–Dawley rats (275–300 g) were obtained from Charles River Laboratories (Hollister, CA) and individually housed, with food and water ad libitum, under a standard 12-h light/12-h dark cycle (lights on 0700–1900 h) at a temperature of 22°C. The protocol for this research was approved by the Institutional Animal Care and Use Committee of the University of California, Irvine, CA. Acquisition, maintenance, handling, procedures, and care of the animals were in accord with the NIH Guide for the Care and Use of Laboratory Animals (NIH Guide, vol. 25, no. 28, 1996).
Drug Treatments
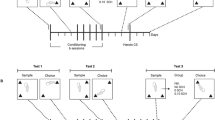
Rats were treated with amphetamines in two separate experiments. In Experiment 1, rats were treated subcutaneously with mAMPH (four injections of 4.0 mg/kg, N=18; three of 21 animals succumbed to complications associated with hyperthermia) or saline (1 ml/kg, N=13), at 2-h intervals. Animals' rectal temperature was measured 1 h after each injection. In Experiment 2, rats were treated subcutaneously with PCA (one injection of 5 mg/kg, N=17), d-AMPH sulfate (d-AMPH) (four injections of 5 mg/kg separated by 2 h, N=20; nine of 29 animals succumbed to complications associated with hyperthermia), or saline (N=16). Rectal temperatures were checked 1, 3, 5, and 7 h following the single injection of PCA, and 1 h following each injection of d-AMPH. All drug and vehicle injections were administered at a volume of 1 ml/kg. All drug doses were chosen based on their known profiles of neurotoxicity, and are expressed as the free base. All drugs were obtained from Sigma Chemical Company (St Louis, MO).
Behavioral Procedures
At 1 week after saline or drug treatment, the animals were exposed to a novelty preference task of OR (Myhrer, 1988; Mumby and Pinel, 1994; Tang et al, 1999). The OR task required that the rats recall which of two small objects they had previously been exposed to. The task took place in a Plexiglas open field (40 × 40 × 38 cm3 high), the outside walls of which were covered with contact paper. A 15 W lamp placed 30 cm above the apparatus provided the only illumination in the room. The Familiarization phase was conducted by placing individual rats for 3 min into the field, in which two identical objects (objects A1 and A2; light bulbs) were positioned in two adjacent corners, 10 cm from the walls (Familiarization phase). In a short-term memory (STM) test given 90 min after familiarization, the rats explored the open field for 3 min in the presence of one familiar (A) and one novel (B; a hook) object. Objects were chosen after determining, in preliminary experiments, that they were equally preferred. Between each trial both the open-field arena and the objects were washed with 95% ethanol solution. All sessions were videotaped, and an experimenter blind to treatment condition analyzed the OR behavior. Exploration was defined as sniffing or touching the object with the nose; sitting on the object was not considered exploration. Object placement was counterbalanced so that half of the animals in each treatment group saw the novel object on the left side (relative to the animal's starting position) of the open-field arena, and the other half saw the novel object on the right side of the arena. The proportion of the total exploration time that the animal spent investigating the novel object was the index of recognition memory for the STM test. An exploration quotient (EQ) calculated for each animal was expressed by the ratio TB/(TA+TB) (TA=time spent exploring the object A; TB=time spent exploring the object B). The same formula was applied in order to get an EQ for the Familiarization phase, with numerator assignment dependent on the side of the novel object during the STM test.
Quantification of 5-HT and Dopamine (DA) Transporters
At 24 h after completion of the last behavioral test, the rats were killed, and their brains were removed and frozen at −20°C for use in autoradiography. In all, 25-μm-thick coronal sections were cut in a cryostat at the level of the striatum and dorsal hippocampus (HC), and sections were incubated with 21 pM [125I]RTI-55 for autoradiographic localization of DA transporters (DAT) in the striatum and 5-HT transporter (SERT) in the HC and perirhinal (pRh) cortex, using the procedures of Boja et al (1992). DAT binding in these limbic areas (HC and pRH cortex) is quite low, and constitutes a small percentage of the total binding (Tohyama and Takatsuji, 1998; Ritchand et al, 1995; Donnan et al, 1989). Similarly, 5-HT innervation comprises only about 20% of the striatal dopaminergic input to the striatum (Brownstein and Palkovits, 1984). For these reasons, only DAT in striatum and SERT in HC and pRh cortex were assessed. For striatal sections, DAT binding was defined as the total amount of [125I]RTI-55 binding in the presence of the SERT inhibitor, fluoxetine (100 nM). For hippocampal (HC) and pRh sections, SERT binding was defined as the total amount of [125I]RTI-55 binding. Slides containing tissue sections and standard slides containing known amounts of radioactivity were apposed to RayMax β autoradiography film (ICN Pharmaceuticals) for 48 h before development. Quantification of [125I]RTI-55 binding to DAT and SERT in the autoradiographs was carried out on an MCID image analyzer (Imaging Research, St Catherines, Ontario, Canada). Image densities were converted to [125I]RTI-55 binding levels using a calibration curve based on images of the standard slides packed with each film. HC and pRh SERT and striatal DAT levels were determined by outlining these structures (based on Paxinos and Watson, 1998) on their respective [125I]RTI-55 images. The regions were quantified for the left and right sides, and readings were averaged from at least three sections per animal. The HC and pRh cortex were chosen because of their known involvement in tasks of learning and memory including OR (see reviews by Squire and Zola-Morgan, 1991; Brown and Aggleton, 2001).
F-J-B Labeling of Cells in Parietal Cortex
For both experiments, a subset of the animals was not exposed to the behavioral task, but was instead killed and perfused 3 days following drug treatment in order to identify degenerating cortical cell bodies following exposure to each of the three amphetamines. Animals were treated with a neurotoxic regimen of mAMPH (N=6), PCA (N=5), or d-AMPH (N=7; two of nine animals succumbed to complications associated with hyperthermia), or they were treated with saline (N=2). This time point (3 days) was chosen based on the observations that the number of F-J positive (F-J+) neurons in the parietal cortex peaked 1–3 days after mAMPH treatment (Eisch et al, 1998; O'Dell and Marshall, 2004). Rats were deeply anesthetized with sodium pentobarbital, then transcardially perfused with PBS (0.1 M phosphate buffer, pH 7.4 with 0.9% NaCl, 50–60 ml), followed by ice-cold 4% paraformaldehyde in PBS (100–120 ml). The perfused brains were removed and postfixed overnight in cold 4% paraformaldehyde in PBS, and transferred to 30% sucrose in 0.1 M PBS at 4°C for 48 h. After cryoprotection in sucrose, 40-μm-thick coronal sections were cut on a freezing microtome and stored in PBS containing sodium azide (1 mg/ml) at 4°C until mounted. All sections to be stained were removed from PBS and incubated in distilled water for 10 min before being mounted onto Vectabond-treated glass slides and allowed to air-dry overnight.
Parietal cortex is the only area in which this laboratory has reliably observed cortical cell degeneration following mAMPH administration (Eisch et al, 1998; O'Dell and Marshall, 2004), although other researchers have obtained evidence of cortical cell degeneration in limbic areas (specifically, piriform cortex, posterolateral cortical amygdaloid nucleus (Bowyer, 2000; Bowyer et al, 2004) and the HC (Schmued and Bowyer, 1997)). Parietal cortex neurons damaged by mAMPH were visualized by staining with F-J, using the procedures developed by Schmued and Hopkins (2000). The slides were initially incubated in ethanol (100% ethanol (3 min), 70% ethanol (1 min)) to help reduce background F-J labeling, and then rinsed in distilled water (2 × 1 min). The slides were then incubated in 0.06% KMnO4 in distilled water (10 min, with moderate agitation) to intensify the F-J labeling of damaged cells and to further reduce background staining. After a distilled water rinse (2 min), the slides were incubated in 0.001% F-J in 0.1% acetic acid for 20 min. Stained slides were finally rinsed in distilled water (3 × 1 min), and then dried on a 45°C slide warmer under a stream of warm air. Dried slides were briefly dipped in xylene, then coverslipped with Fluoromount. F-J- stained sections were examined by epifluorescence using a mercury–helium lamp with a blue excitation filter set (Nikon, BA-2: excitation filter=450–490 nm; dichroic mirror=505 nm; emission filter=520 nm barrier). Three sections per animal within the region of parietal cortex we have previously found susceptible to mAMPH (PAR1; Eisch et al, 1998; Eisch and Marshall, 1998) were examined.
Statistical Analysis
Object recognition EQs between groups were compared using analysis of variance (ANOVA). Within-group comparisons of EQs between the familiarization phase and test session were carried out with paired t-tests. [125I]RTI-55 binding results were analyzed by ANOVA. F-J+ cells in parietal cortex were visualized by microscopic examination and counted manually. Data were summed across hemispheres for three sections per animal and reported as total number of labeled cells/animal. Bonferroni correction was used for post hoc treatment group comparisons.
RESULTS
OR Behavior
mAMPH induced impairments in STM for novel objects
Three animals died from hyperthermia due to mAMPH treatments. During the treatments with either mAMPH or saline, the animals' core body temperatures were measured, as described in the Materials and Methods section. Animals treated with mAMPH had significantly higher peak body temperatures (40.5±0.2°C) than did saline-treated controls (38.3±0.2°C; Independent samples t-test, t=9.885, p<0.001). Additionally, mAMPH-treated animals had significantly higher core body temperatures at each of the four 1-h postinjection temperature readings, compared to saline-injected controls (Table 1).
Animals exposed to mAMPH or saline showed no differences 1 week later in the Familiarization phase of the task, with both groups spending equivalent amounts of time exploring the two objects (the sum of time spent investigating objects A1 and A2) (independent-samples t-test, t=0.322, p>0.05). Additionally, both the mAMPH and saline-treated groups' investigatory behavior of the two identical objects was equivalent, with both groups showing EQs around 50% during the familiarization phase (object A1 vs object A2 paired-samples t-test, t=0.494, p>0.05 mAMPH, and t=0.901, p>0.05, saline). These findings indicate that (1) mAMPH treatment does not influence object exploration during the Familiarization phase, and (2) both copies of the object are equivalently preferred.
In the STM test given 90 min later, one of the familiar objects (lightbulb) was replaced by a novel object (hook). A repeated measures ANOVA revealed a significant interaction between the groups on their percent of time spent investigating the objects (EQ scores) at the Familiarization phase compared to the STM test (F(1, 26)=7.477, p=0.011). Saline control animals showed a strong preference towards investigating the novel object, indicated by a high EQ on the STM test (Familiarization phase EQ vs STM EQ, paired samples t-test, p=0.014), whereas mAMPH-treated animals did not, indicating that prior treatment with mAMPH blocked memory retention for the familiar object (Familiarization phase EQ vs STM EQ, paired-samples t-test, p=0.413) (Figure 1). Thus, although the groups showed equivalent object exploration during the familiarization phase, the administration of mAMPH produced profound deficits in performance during the STM OR test.

Methamphetamine (mAMPH) impairs STM performance in the novel OR memory task 1 week after drug treatment. (See Materials and methods for definition of the EQ.) Data expressed as mean±SEM. *mAMPH-treated group differs from corresponding saline-treated control group, p=0.01.
Effects of PCA and d-AMPH on STM for novel objects
Treatment with either PCA or d-AMPH caused a significant increase in the animals' peak body core temperature relative to saline-treated control animals (one-way ANOVA, F(2, 41)=45.192, p<0.001). Additionally, multiple comparisons revealed that each of the groups was significantly different from the others (Bonferroni correction, p<0.001 for all group comparisons), with d-AMPH-treated animals having the highest average peak temperature (40.7±0.6°C) relative to PCA-treated (39.5±0.7°C) and saline-treated animals (38.8±0.3°C). Each of the groups' average temperatures across the four temperature readings is shown in Table 1.
Animals treated with either PCA or d-AMPH showed no differences 1 week later in their overall object exploration times in the Familiarization phase of the OR task, compared to saline controls (one-way ANOVA, F(2, 43)=1.007, p>0.05). Additionally, each of the groups' investigation of the two identical objects was equivalent (paired-samples t-test, PCA: t=0.0203; d-AMPH: t=0.649; saline: t=0.195, p>0.05 for all tests).
In this experiment, there was a significant effect of test phase but no treatment by test interactions (3 × 2 repeated measures ANOVA, F(1, 41)=36.862, p=0.000 and F(2, 41)=0.611, p>0.05, respectively). Animals treated with PCA, d-AMPH or saline showed a significant preference for the novel object in the STM session, indicated by a higher EQ at the STM test than during the Familiarization phase (Familiarization phase EQ vs STM EQ, paired-samples t-tests; p=0.001, 0.002, and 0.009 for PCA, d-AMPH, and saline-treated animals, respectively). These results indicate that exposure to either PCA or d-AMPH did not impair performance on a novelty preference task of OR memory (Figure 2).

Animals subjected to neurotoxic regimens of either p-chloroamphetamine (PCA) or d-amphetamine (d-AMPH) showed no differences from saline-treated controls when tested on the novelty preference test of OR 1 week later. Data expressed as mean±SEM.
DAT and SERT Binding
Loss of DAT and SERT binding sites in the brains of mAMPH-treated rats
To assess mAMPH-induced damage to the dopaminergic and serotonergic terminals in diverse areas of the brain, we used autoradiography to quantify radioligand binding to DAT and SERT in the brains of animals given mAMPH or saline and that were used in the OR test 1 week after treatment. mAMPH treatment induced significant depletions (approximately 41%) in levels of binding to DAT in the ventral caudate putamen (multivariate ANOVA, F(1, 26)=17.436, p<0.001), and a slight, nonsignificant 20% decrease in DAT binding in the dorsal caudate putamen (F(1, 26)=4.171, p=0.051). DAT binding in the nucleus accumbens was unaffected (Figure 3). Treatment with mAMPH also caused a significant decrease in levels of RTI binding to SERT in the HC (an approximate 23% decrease; multivariate ANOVA, F(1, 26)=9.919, p=0.004) and pRh cortex (a decrease of about 21%; F(1, 26)=7.332, p=0.012) (Figures 3 and 5). Also apparent from Figure 5 is that mAMPH also induced a loss of SERT binding in other brain regions (eg neocortex).

RTI binding to dopaminergic transporters (DAT) in dorsal (dCPu) and ventral caudate-putamen (vCPu) and nucleus accumbens (NAc), and to serotonergic transporters (SERT) in hippocampus (HC) and perirhinal cortex (pRh Cx). Values represent mean binding values, expressed as μCi/g of tissue. *Denotes difference between saline- and methamphetamine-treated groups, p<0.01.

Dopamine transporter (DAT) and serotonin transporter (SERT) binding sites in brains of saline, methamphetamine (mAMPH)-, p-chloroamphetamine (PCA)-, or d-amphetamine (d-AMPH)-treated animals. Autoradiographic images of [125I]RTI-55 binding to striatal DAT (top row), hippocampal SERT (middle row), and perirhinal SERT (bottom row) of rats used in the behavioral tasks 2 weeks after saline (a, e, i), mAMPH (b, f, j), PCA (c, g, k), or d-AMPH (d, h, l) injections.
Loss of SERT and DAT binding sites in the brains of PCA- and amphetamine-treated animals, respectively
Animals treated with PCA showed a highly significant loss of RTI binding to serotonergic transporter proteins in the HC (64%; F(2, 41)=60.034, p<0.0001) and pRh cortex (87%; F(2, 41)=185.803, p<0.0001), whereas dopaminergic terminals in nucleus accumbens, as well as dorsal and ventral caudate-putamen were intact (Figures 4 and 5). As is apparent from Figure 5, binding to SERT was also affected in other brain regions (eg neocortex).

RTI binding to dopaminergic (DAT) or serotonergic (SERT) transporters in the dorsal (dCPu) and ventral caudate putamen (vCPu), nucleus accumbens (NAc), hippocampus (HC), and perirhinal cortex (pRh Cx). Values represent mean binding levels, expressed in μCi/g of tissue. Differs from corresponding saline-treated group: ap<0.01; bp<0.001.
Animals treated with d-AMPH showed highly significant loss of RTI binding to dopaminergic transporters in three subregions of the striatum, the dorsal caudate–putamen (45%; F(2, 41)=18.829, p<0.0001), ventral caudate–putamen (65%; F(2, 41)=33.426, p<0.0001), and to a lesser degree, the nucleus accumbens (25%; F(2, 41)=5.376, p<0.01). However, serotonergic terminals were spared from damage following d-AMPH treatment (Figures 4 and 5).
F-J-B Labeling of Degenerating Cortical Neurons
In all, 20 animals not used for OR testing were employed to quantify cortical cell degeneration. As the absolute numbers of parietal cortex F-J+ cells were quite variable within the treatment groups, a minimum criterion of 10 F-J+ cells (per three sections examined) was set in order for an animal to be counted as having significant cortical cell degeneration. Using this criterion, 100% of the animals treated with either mAMPH (median number of F-J+ cells=51, range=19–183) or d-AMPH (median=22, range=20–597) showed F-J+ cell degeneration in the barrel field of the primary somatosensory cortex. By this same criterion, 0% of the PCA- (median=1, range=0–2) and saline-treated animals (median=0, range=0) showed F-J labeling.
DISCUSSION
In sufficient doses, mAMPH causes long-term damage to DA terminals in the striatum and to forebrain 5-HT terminals, as evidence by long-term depletions in these two neurotransmitters, their synthetic enzymes, metabolites, and plasma membrane transporters (Koda and Gibb, 1973; Hotchkiss and Gibb, 1980; Schmidt et al, 1985; Wagner et al, 1979; Ricaurte et al, 1982; Axt and Molliver, 1991; Friedman et al, 1998; Chapman et al, 2001). The mAMPH-induced depletions of DA reverse with a very slow time course (Cass and Manning, 1999). Additionally, this regimen causes degeneration of neurons in somatosensory cortex (Commins and Seiden, 1986; Eisch et al, 1998; O'Dell and Marshall, 2000). The dopaminergic loss has been interpreted as neurotoxicity on the basis of both neurochemical and histological evidence (Koda and Gibb, 1973; Fibiger and McGeer, 1971; Seiden et al, 1975; Ricaurte et al, 1982; Volkow et al, 2001a, 2001b; McCann et al, 1998), but the issue of whether mAMPH induces DA terminal degeneration has been challenged by studies of mAMPH-treated animals (Harvey et al, 2000) and human mAMPH abusers (Wilson et al, 1996; Volkow et al, 2001a, 2001b). Whether or not the decreases in markers of DA and 5-HT terminals constitute terminal degeneration or a long-lasting phenotypic downregulation, the focus of the present study was to determine the cognitive consequences of this loss of monoaminergic markers.
The present findings fit into a larger literature concerning long-term behavioral changes after neurotoxic regimens of mAMPH and other amphetamines. Animals show motor impairments in a balance beam and in active avoidance tasks following a neurotoxic regimen of mAMPH (Walsh and Wagner, 1992). Chapman et al (2001) found impairment in a radial arm maze task of sequential motor learning in rats that were exposed to neurotoxic doses of mAMPH. Friedman et al (1998) report a mild impairment in the acquisition of the Morris Water Maze task of spatial learning, but Schröder et al (2003) reported no impairment in acquisition or retention of this task in mAMPH-treated animals. The current research corroborated findings reported by Bisagno et al (2002) and Schröder et al (2003) that the repeated administration of mAMPH within a single day produces profound deficits in a nonspatial task of recognition memory.
This mAMPH-induced OR impairment could be due to one, or a combination, of the following possibilities: (1) dopaminergic system terminal neurotoxicity, (2) serotonergic system terminal neurotoxicity, (3) cortical cell degeneration, or (4) other effects of mAMPH, some of which may be unknown. The primary purpose of these experiments was to investigate the first two possibilities. Two amphetamine analogues were used that achieve similar acute physiological effects as occur with mAMPH administration (eg increased body core temperature and behavioral activation), but differ in their profiles of CNS neurotoxicity. Animals given a neurotoxic regimen of either PCA (a drug that causes long-lasting depressions in serotonergic, but not dopaminergic terminal markers), or d-AMPH (a drug that causes enduring losses in markers of dopaminergic, but not serotonergic terminals), were unimpaired in the task of OR.
Although there is a large body of research investigating the PCA-induced impairments, most of these studies examined behavioral changes immediately after PCA administration (Solana-Figueroa et al, 2002; Archer et al, 1982; Ögren, 1982). Ruotsalainen et al (2000) report mild impairments in response inhibition in animals 3 weeks following a neurotoxic regimen of PCA, but no impairment in a 5-choice serial reaction time task of attention. We are unaware of any studies showing lasting memory impairments in animals following 5-HT depletion by PCA.
Another amphetamine analogue, 3,4-methylenedioxymethamphetamine (MDMA), is a popular recreational drug of abuse and, like PCA, has structural similarities to mAMPH and exerts greater neurotoxic effects on serotonergic than to dopaminergic systems (McCann and Ricaurte, 1994; Lyles and Cadet, 2003). Rats given neurotoxic doses of MDMA were mildly impaired in developing efficient search strategies in a place navigation task (Robinson et al, 1993). MDMA also produced deficits in a delayed non-match-to-place task in rats (Marston et al, 1999). Morley et al (2001) report that rats exposed to high (but not low) doses of MDMA showed impairments on the novelty preference OR task. However, it is presently unknown whether neurotoxic damage to 5-HT systems was responsible for the cognitive impairment observed by Morley et al (2001).
To assess the possibility that the mAMPH-induced OR impairment could be due to the mAMPH-induced damage to cortical cells in somatosensory cortex, fluorescence histochemistry was used to qualify cortical cell degeneration following exposure to mAMPH, PCA, or d-AMPH. PCA-treated animals did not show significant cell loss in this region of cortex, whereas mAMPH and d-AMPH-treated animals did. Behaviorally, mAMPH-treated animals exhibited OR impairments, but both d-AMPH- and PCA-treated animals exhibited no deficits on the task, making it unlikely that injury to cells in somatosensory cortex were responsible for the cognitive impairment.
Administration of mAMPH, PCA, or d-AMPH induces a significant increase in body core temperature. Although we do not assume that hyperthermia by itself is responsible for the mAMPH-induced cognitive impairment, it is possible that a confluence of several mAMPH-induced factors causes impairment in OR behavior, and that one of these responsible events includes an increase in body temperature. However, this possibility appears unlikely because both PCA and d-AMPH raised body core temperature without impairing OR.
The findings reported in this study appear to indicate that damage to either DA or 5-HT alone is insufficient to produce the impairments seen in OR following a neurotoxic mAMPH regimen. However, using other brain lesion approaches, several authors report altered behavioral function following partial, but not complete, damage to brain structures in rodents (Solomon et al, 1983), non-human primates (Meunier et al, 1996; Baxter and Murray, 2001) and humans (Mayes et al, 2002), implying an inverse correlation between lesion size and intact cognitive function. While several theories have been offered to explain this seemingly paradoxical finding, one of the most intriguing hypotheses is that, following incomplete structural lesion, task functions that would otherwise be subsumed by surrounding structures receives competition from remaining yet compromised circuits (see Baxter and Murray, 2001 for a more comprehensive discussion). Given that the PCA- and d-AMPH-induced effects on SERT and DAT, respectively, are both greater in magnitude than those observed following mAMPH administration, we cannot exclude the possibility that differences between the relative extents of damage caused by each amphetamine analogue contribute to the distinct behavioral results in the task of OR.
Alternatively, it may be that both the DA and 5-HT systems must be compromised for these cognitive impairments to occur, and it remains to be determined whether damaging both serotonergic and dopaminergic terminals (by combining PCA and d-AMPH, for example) would yield impairments in OR memory. Finally, although much is known about the effects of a neurotoxic mAMPH regimen on monoaminergic terminals, this amphetamine may exact effects on other neural systems, which have not yet been reported, but could play an integral role in the long-term cognitive effects of this drug of abuse. Future studies are needed to explore these possibilities and uncover the reason for the reported memory impairment following mAMPH neurotoxicity. Such studies could provide important information in the treatment of cognitive disorders in the population of stimulant drug abusers.
References
Archer T, Ogren SO, Ross SB (1982). Serotonin involvement in aversive conditioning: reversal of the fear retention deficit by long-term pichloroamphetamine but not p-chlorophenylalanine. Neurosc Lett 34: 75–82.
Axt KJ, Molliver ME (1991). Immunocytochemical evidence for methamphetamine-induced serotonergic axon loss in the rat brain. Synapse 9: 302–313.
Baxter MG, Murray EA (2001). Opposite relationship of hippocampal and rhinal cortex damage to delayed nonmatching-to-sample deficits in monkeys. Hippocampus 11: 61–71.
Bisagno V, Ferguson D, Luine VN (2002). Short toxic methamphetamine schedule impairs object recognition task in male rats. Brain Res 940: 95–101.
Boja JW, Mitchell WM, Patel A, Kopajtic TA, Carroll FI, Lewin AH et al (1992). High-affinity binding of [125I]RTI-55 to dopamine and serotonin transporters in rat brain. Synapse 12: 27–36.
Bowyer JF (2000). Neuronal degeneration in the limbic system of weanling rats exposed to saline, hyperthermia or d-amphetamine. Brain Res 885: 166–171.
Bowyer JF, Delongchamp RR, Jakab RL (2004). Glutamate N-methyl-D-aspartate and dopamine receptors have contrasting effects on the limbic versus the somatosensory cortex with respect to amphetamine-induced neurodegeneration. Brain Res 1030: 234–246.
Bowyer JF, Peterson SI, Rountree RL, Tor-Agbidye J, Wang GJ (1998). Neuronal degeneration in rat forebrain resulting from D-amphetamine-induced convulsions is dependent on seizure severity and age. Brain Res 809: 77–90.
Brown MW, Aggleton JP (2001). Recognition memory: what are the roles of the perirhinal cortex and hippocampus? Nat Rev Neurosci 2: 51–61.
Brownstein MJ, Palkovits M (1984). Catecholamines, serotonin, acetylcholine and γ-aminobutyric acid in the rat brain: biochemical studies. In: Björklund A, Hökfelt T (eds). Handbook of Chemical Neuroanatomy: Volume 2: Classical Transmitters in the CNS. Part I. Elsevier: New York. pp 23–54.
Cass WA, Manning MW (1999). Recovery of presynaptic dopaminergic functioning in rats treated with neurotoxic doses of methamphetamine. J Neurosc 19: 7653–7660.
Chapman DE, Hanson GR, Kesner RP, Keefe KA (2001). Long-term changes in basal ganglia function after a neurotoxic regimen of methamphetamine. J Pharmacol Exp Therap 296: 520–527.
Commins DL, Seiden LS (1986). Alpha-methyltyrosine blocks methylamphetamine-induced degeneration in the rat somatosensory cortex. Brain Res 365: 15–20.
Donnan GA, Kaczmarczyk SJ, McKenzie JS, Kalnins RM, Chilco PJ, Mendelsohn FAO (1989). Catecholamine uptake sites in mouse brain: distribution determined by quantitative [3H]mazindol autoradiography. Brain Res 504: 64–71.
Eisch AJ, Gaffney M, Weihmuller FB, O'Dell SJ, Marshall JF (1992). Striatal subregions are differentially vulnerable to the neurotoxic effects of methamphetamine. Brain Res 598: 321–326.
Eisch AJ, Marshall JF (1998). Striatal and cortical NMDA receptors are altered by a neurotoxic regimen of methamphetamine. Synapse 22: 217–225.
Eisch AJ, Schmued LC, Marshall JF (1998). Characterizing cortical neuron injury with Fluoro-Jade labeling after a neurotoxic regimen of methamphetamine. Synapse 30: 329–333.
Ennaceur A, Delacour J (1988). A new one-trial test for neurobiological studies of memory in rats. 1: behavioral data. Behavioral Brain Res 31: 47–59.
Fibiger HC, McGeer PL (1971). Effect of acute and chronic methamphetamine treatment on tyrosine hydroxylase activity in brain and adrenal medulla. Eur J Pharmacol 16: 176–180.
Friedman SD, Castaneda E, Hodge GK (1998). Long-term monoamine depletion, differential recovery, and subtle behavioral impairment following methamphetamine-induced neurotoxicity. Pharmacol Biochem Behav 61: 35–44.
Fuller RW, Hines CW, Mills J (1965). Lowering of brain serotonin level by chloroamphetamines. Biochem Pharmacol 14: 483–488.
Harvey DC, Lacan G, Tanious SP, Melega WP (2000). Recovery from methamphetamine induced long-term nigrostriatal dopaminergic deficits without substantia nigra cell loss. Brain Res 871: 259–270.
Harvey JA, McMaster SE, Yunger LM (1975). p-Chloroamphetamine: Selective neurotoxic action in brain. Science 187: 841–843.
Hotchkiss AJ, Gibb JW (1980). Long term effects of multiple doses of methamphetamine on tryptophan hydroxylase and tyrosine hydroxylase activity in rat brain. J Pharmacol Exp Therap 214: 257–262.
Koda LY, Gibb JW (1973). Adrenal and striatal tyrosine hydroxylase activity after methamphetamine. J Pharmacol Exp Therap 185: 42–48.
Lyles J, Cadet JL (2003). Methylenedioxymethamphetamine (MDMA, Ecstasy) neurotoxicity: cellular ad molecular mechanisms. Brain Res Rev 42: 155–168.
Marston HM, Reid ME, Lawrence JA, Olverman HJ, Butcher SP (1999). Behavioral analysis of the acute and chronic effects of MDMA treatment in the rat. Psychopharmacology 144: 67–76.
Mayes AR, Holdstock JS, Isaac CL, Hunkin NM, Roberts N (2002). Relative sparing of item recognition memory in a patient with adult-onset damage limited to the hippocampus. Hippocampus 2: 325–340.
McCann UD, Ricaurte GA (1994). Use and abuse of ring-substituted amphetamines. In: Cho AK, Segal DS (eds). Amphetamine and Its Analogs. Academic Press: San Diego. pp 371–386.
McCann UD, Wong DF, Yokoi F, Villemagne V, Dannals RF, Ricaurte GA (1998). Reduced striatal dopamine transporter density in abstinent methamphetamine and methcathinone users: evidence from positron emission tomography studies with [11C]WIN-35 428. J Neurosc 18: 8417–8422.
Meunier M, Hadfield W, Bachevalier J, Murray EA (1996). Effects of rhinal cortex lesions combined with hippocampectomy on visual recognition memory in rhesus monkeys. J Neurophysiol 75: 1190–1205.
Morley KC, Gallate JE, Hunt GE, Mallet PE, McGregor IS (2001). Increased anxiety and impaired memory in rats 3 months after administration of 3,4-methylenedioxymethamphetamine (‘Ecstasy’). Eur J Pharmacol 433: 91–99.
Mumby DG, Pinel JP (1994). Rhinal cortex lesions and object recognition in rats. Behav Neurosci 108: 11–18.
Myhrer T (1988). Exploratory behavior and reaction to novelty in rats with hippocampal perforant path systems disrupted. Behav Neurosci 102: 356–362.
O'Dell SJ, Marshall JF (2000). Repeated administration of methamphetamine damages cells in the somatosensory cortex: overlap with cytochrome oxidase-rich barrels. Synapse 37: 32–37.
O'Dell SJ, Marshall JF (2004). Neurotoxic regimens of methamphetamine induce persistent expression of phospho-c-Jun in somatosensory cortex and substantia nigra. Synapse 55: 137–147.
Ögren SO (1982). Forebrain serotonin and avoidance learning: behavioural and biochemical studies on the acute effect of p-chloroamphetamine on one-way active avoidance learning in the male rat. Pharmacol Biochem Behav 16: 881–895.
Paxinos G, Watson C (1998). The Rat Brain in Stereotaxic Coordinates. Academic Press: San Diego.
Pletscher A, Burkard WP, Bruderer H, Gey KF (1963). Decrease of cerebral 5-hydroxytryptamine and 5-hydroxyindoleacetic acid by arylalklamine. Life Sci 11: 828–833.
Ricaurte GA, Guillery RW, Seiden LS, Schuster CR, Moore RY (1982). Dopamine nerve terminal degeneration produced by high doses of methylamphetamine in the rat brain. Brain Res 2–35: 93–103.
Ricaurte GA, Schuster CR, Seiden LS (1980). Long-lasting effects of repeated methamphetamine administration on dopamine and serotonin neurons in the rat brain: a regional study. Brain Res 193: 153–160.
Ritchand NM, Kelsoe JR, Segal DS, Kuczenski R (1995). Regional quantification of dopamine transporter mRNA in rat brain using a ribonuclease protection assay. Neurosci Lett 200: 73–76.
Robinson TE, Castenada E, Wishaw IQ (1993). Effects of cortical serotonin depletion by 3,4-methylenedioxymethamphetamine (MDMA) on behavior, before, and after additional cholinergic blockade. Neuropsychopharmacology 8: 77–85.
Ruotsalainen S, Miettinen R, MacDonald E, Koivisto E, Sirvio J (2000). Blockade of muscarinic, rather than nicotinic, receptors impairs attention, but does not interact with serotonin depletion. Psychopharmacology (Berl) 148: 111–123.
Sanders-Bush E, Bushing JA, Sulser F (1972). Long-term effects of p-chloroamphetamine on tryptophan hydroxylase activity and on the levels of 5-hydroxytryptamine and 5-hydroxyindoleacetic acid in brain. Eur J Pharmacol 20: 385–388.
Schmidt CJ, Ritter JK, Sonsalla PK, Hanson GR, Gibb JW (1985). Role of dopamine in the neurotoxic effects of methamphetamine. J Pharmacol Exp Therap 233: 539–544.
Schmued LC, Albertson C, Slikker Jr W (1997). Fluoro-Jade: a novel fluorochrome for the sensitive and reliable histochemical localization of neuronal degeneration. Brain Res 751: 37–46.
Schmued LC, Bowyer JF (1997). Methamphetamine exposure can produce neuronal degeneration in mouse hippocampal remnants. Brain Res 759: 135–140.
Schmued LC, Hopkins KJ (2000). Fluoro-Jade B: a high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res 874: 123–130.
Schröder N, O'Dell SJ, Marshall JF (2003). Neurotoxic methamphetamine regimen severely impairs recognition memory in rats. Synapse 49: 89–96.
Seiden LS, Fischman MW, Schuster CR (1975). Long-term methamphetamine induced changes in brain catecholamines in tolerant rhesus monkeys. Drug Alcohol Depen 1: 215–219.
Simon SL, Domier CP, Sim T, Richardson K, Rawson RA, Ling W (2002). Cognitive performance of current methamphetamine and cocaine abusers. J Addict Dis 21: 61–74.
Solana-Figueroa R, Salado-Castillo R, Galindo LE, Quirarte GL, Prado-Alcala RA (2002). Effects of pretraining intrastriatal administration of p-chloroamphetamine on inhibitory avoidance. Neurobiol Learning Mem 78: 178–185.
Solomon PR, Solomon SD, Schaaf EV, Perry HE (1983). Altered activity in the hippocampus is more detrimental to classical conditioning than removing the structure. Science 220: 329–331.
Squire LR, Zola-Morgan S (1991). The medial temporal lobe memory system. Science 253: 1380–1386.
Tang YP, Shimizu E, Dube GR, Rampon C, Kerchner GA, Zhuo M et al (1999). Genetic enhancement of learning and memory in mice. Nature 401: 63–69.
Tohyama M, Takatsuji K (1998). Atlas of Neuroactive Substances and Their Receptors in the Rat. Oxford University Press: Oxford.
Volkow ND, Chang L, Wang GJ, Fowler JS, Franceschi D, Sedler M et al (2001a). Loss of dopamine transporters in methamphetamine abusers recovers with protracted abstinence. J Neurosci 21: 9414–9418.
Volkow ND, Chang L, Wang GJ, Fowler JS, Leonido-Yee M, Franceschi D et al (2001b). Association of dopamine transporter reduction with psychomotor impairment in methamphetamine abusers. Am J Psychiatry 158: 377–382.
Wagner GC, Ricaurte GA, Seiden LS, Schuster CR, Miller RJ, Westley J (1980). Long-lasting depletions of striatal dopamine and loss of dopamine uptake sites following repeated administration of methamphetamine. Brain Res 181: 151–160.
Wagner GC, Seiden LS, Schuster CR (1979). Methamphetamine-induced changes in brain catecholamines in rats and guinea pigs. Drug Alcohol Depen 4: 435–438.
Walsh SL, Wagner GC (1992). Motor impairments after methamphetamine-induced neurotoxicity in the rat. J Pharmacol Exp Therap 263: 617–626.
Wilson JM, Kalasinsky KS, Levey AI, Bergeron C, Reiber G, Anthony RM et al (1996). Striatal dopamine nerve terminal markers in human, chronic methamphetamine users. Nat Med 2: 699–703.
Acknowledgements
This research was supported by PHS Grant DA 05538. We thank Bryan Casiano and Thuy Nguyen for technical assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Belcher, A., O'Dell, S. & Marshall, J. Impaired Object Recognition Memory Following Methamphetamine, but not p-Chloroamphetamine- or d-Amphetamine-Induced Neurotoxicity. Neuropsychopharmacol 30, 2026–2034 (2005). https://doi.org/10.1038/sj.npp.1300771
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.npp.1300771
Keywords
This article is cited by
-
Neurochemical and behavioral comparisons of contingent and non-contingent methamphetamine exposure following binge or yoked long-access self-administration paradigms
Psychopharmacology (2020)
-
Effect of three different regimens of repeated methamphetamine on rats’ cognitive performance
Cognitive Processing (2018)
-
Current understanding of methamphetamine-associated dopaminergic neurodegeneration and psychotoxic behaviors
Archives of Pharmacal Research (2017)
-
Sargassum swartzii extracts ameliorate memory functions by neurochemical modulation in a rat model
Food Science and Biotechnology (2017)
-
Methamphetamine-, d-Amphetamine-, and p-Chloroamphetamine-Induced Neurotoxicity Differentially Effect Impulsive Responding on the Stop-Signal Task in Rats
Neurotoxicity Research (2016)
