
Abstract
Although considerable progress has been made towards understanding the neural systems mediating conditioned fear, little is known about the neural mechanisms underlying conditioned inhibitors of fear (or safety signals). The present series of experiments examined the involvement of the nucleus accumbens (NAC) in mediating the effects of safety signals on behavior using a conditioned inhibition of fear-potentiated startle paradigm. Neither increasing dopaminergic nor decreasing glutamatergic function in the NAC altered the magnitude of conditioned fear or conditioned inhibition of fear in rats. Furthermore, large pre- or post-training electrolytic lesions of the NAC did not affect acquisition or expression of fear-potentiated startle or conditioned inhibition of fear-potentiated startle. Taken together, these data suggest that the NAC is not critically involved in the acquisition or expression of fear-potentiated startle or conditioned inhibition of fear-potentiated startle. Previous research has implicated the NAC in ‘reward-attenuated startle’ in which presentation of a stimulus paired with food decreased startle responding. The present results, therefore, indicate important neural dissociations between the processing of appetitive and safety signals, even though behavioral studies and learning theories have suggested that these two forms of learning share some commonalities.
Similar content being viewed by others


Introduction
Classical fear conditioning has been used extensively to delineate the neural systems mediating conditioned fear. In these experiments, a previously neutral stimulus (eg, light or tone; conditioned stimulus; CS) that is paired with an aversive stimulus (footshock; unconditioned stimulus; US) acquires the ability to elicit conditioned fear responses. Results from many labs using this task show that the acquisition and expression of Pavlovian fear conditioning critically involves the amygdala (for reviews, see Davis, 1992; LeDoux, 2000; Schafe et al, 2001; Wallace and Rosen, 2001; Fanselow and Gale, 2003; Maren, 2003). Once acquired, conditioned fear may last a lifetime (Fanselow and Gale, 2003; Gale et al, 2004). However, the expression of conditioned fear acquired under one set of circumstances may be inappropriate or even disadvantageous in another set of circumstances in which the CS no longer predicts a realistic threat. Therefore, it is also important to understand the neural mechanisms underlying the process of fear inhibition or reduction. The findings from these studies may provide therapeutic insights into clinical states associated with dysfunctions of fear inhibition such as anxiety disorders.
Fear may be reduced or inhibited in several ways. For example, extinction training, in which a stimulus that was previously paired with shock is presented repeatedly in the absence of the shock (CS-no shock), gradually reduces conditioned fear responses (Pavlov, 1927; Cain et al, 2003; Quirk and Gehlert, 2003). Conditioned inhibition of fear, in which a stimulus signals the absence of shock, is another procedure that reduces conditioned fear responses (Rescorla, 1969). In one example of this paradigm, a light is presented with a shock (Light-shock) but, on some trials, a noise is paired with the light in the absence of shock (Noise & Light-no shock). Following several pairings, the light acquires the ability to elicit conditioned fear responses and the noise acquires the ability to reduce the conditioned fear responses elicited by the light. In this way, the noise comes to predict the absence of shock and hence becomes a safety signal (Falls and Davis, 1995).
Little is known about the neural systems that mediate the fear-inhibiting effects of a safety signal. However, several lines of evidence converge to suggest that the nucleus accumbens (NAC) may be involved. First, the NAC receives inputs from many brain regions, including those involved in conditioned fear (McDonald, 1991; Brog et al, 1993). Indeed, descending glutamatergic projections from the amygdala, hippocampus and prefrontal cortex, and ascending dopaminergic fibers arising from the midbrain converge at the NAC (Groenewegen et al, 1999). Second, presentation of a safety signal is associated with an increase in the expression of immediate early genes in the NAC, among other areas (Campeau et al, 1997).
Finally, an extension of some learning theories implicates the involvement of the NAC. These theories hypothesize the existence of two mutually antagonistic motivational systems in the brain; the appetitive and aversive motivational systems (Konorski, 1967; Dickinson and Dearing, 1979). As these motivational systems would reciprocally inhibit one another, activation of the aversive system would inhibit the appetitive system. By extension, a safety signal that inhibits the aversive system may be perceived by the animal as motivationally equivalent to a stimulus that directly activates the appetitive motivational system. There is general agreement that the NAC is critically involved in mediating the motivational impact of appetitive Pavlovian cues on behavior (Robbins et al, 1989; Berridge and Robinson, 1998; Dayan and Balleine, 2002; de Borchgrave et al, 2002). For instance, a stimulus that was previously paired with sucrose availability enhances both instrumental responding for sucrose (an effect known as Pavlovian-to-instrumental transfer) and elicits an increase in locomotion (an effect known as autoshaping) (Brown and Jenkins, 1968). Importantly, both of these effects of are blocked by lesions of the NAC (Balleine and Killcross, 1994; Parkinson et al, 2000; Corbit et al, 2001; Hall et al, 2001) while enhancing dopaminergic transmission in the NAC increases Pavlovian-to-instrumental transfer (Wyvell and Berridge, 2000). Of interest is the recent finding that presentation of a safety signal increases locomotion (Rogan et al, 2003), perhaps by a mechanism similar to autoshaping. It is conceivable, therefore, that the NAC may also mediate the effects of a safety signal on behavior.
The present study, therefore, examined the role of the NAC in mediating the effects of safety signals on behavior using conditioned inhibition of fear-potentiated startle. As dopamine- and glutamate-mediated mechanisms are key to NAC function (Choi et al, 2000; Everitt and Wolf, 2002; David et al, 2004), Experiments 1a and 1b examined the effects of enhancing dopaminergic function (via the dopamine agonist, amphetamine) or disrupting glutamatergic function (via the AMPA/kainate receptor antagonist, CNQX) in the NAC on the expression of conditioned inhibition of fear-potentiated startle. Previous research shows that infusion of amphetamine into the NAC disrupts prepulse inhibition of the acoustic startle response (eg, Swerdlow et al, 1986; Wan et al, 1995). As a positive control to ensure that our infusions produced adequate perfusion of the NAC, Experiment 1c examined the effects of infusing amphetamine into the NAC in the same rats on prepulse inhibition. Experiments 2a and 2b examined the effects of electrolytic lesions of the NAC on the acquisition and retention of conditioned inhibition of fear-potentiated startle, respectively.
Methods and Materials
Animals
Male albino Sprague–Dawley rats (Charles, River, Kingston, NY, USA) weighing between 300–400 g were used (unless otherwise specified). Rats were housed in hanging wire cages (two rats per cage) and maintained on a 24-h light–dark cycle (lights on at 7 am) with ad libitum access to food and water. All procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals, published by the United States Public Health Service, with the experimental protocols approved by Yale University and the Northern Illinois University Institutional Animal Care and Use Committees. All efforts were made to minimize the number of animals used.
Cannulation Procedure and Infusions
Rats (n=15) were anesthetized (sodium pentobarbital, 60 mg/kg, i.p.) and placed in a Kopf stereotaxic instrument. Bilateral chronic indwelling cannula (22 gauge, Plastic One, Roanoke, VA, USA) were implanted into the NAC (co-ordinates Anterior/Posterior=+1.2, Medial/Lateral=±1.2, Dorsal/Ventral=−7.4 relative to bregma, Paxinos and Watson, 1998).
Infusions were performed immediately before testing. Injection cannulas (Plastics One, Model C3131; 28gauge) cut to extend 1 mm beyond the tip of the guide cannulas were attached by polyethylene tubing to Hamilton microsyringes controlled by an infusion pump (Harvard Apparatus, South Natick, MA, USA). The volume of all infusions was 0.5 μl (infused over 60 s). After the infusion was complete, the injection cannula was left in place an additional 60 s to increase diffusion of the drug. We chose to infuse sufficient volume of fluid to perfuse the entire NAC since we had no a priori prediction as to the involvement of a specific NAC subterritory (core, shell and rostral pole (Zahm, 2000), or combination thereof).
Amphetamine (Sigma Chemicals, St Louis, MO) was dissolved in distilled water to result in concentrations of 0, 10, and 20 μg/0.5 μl. Previous experiments show that this dose range of amphetamine increased the motivational impact of appetitive Pavlovian cues on behavior (eg, locomotion and responding for conditioned reinforcements) (Parkinson et al, 1999). The AMPA receptor antagonist, 6-cyanao-7-nitroquinoxaline-2,3-dione (CNQX) (Sigma Chemicals) was dissolved in 1 N NaOH and diluted with 0.1 M PBS. The pH of the resulting solution was adjusted to 7.4. Previous studies show that the present dose of CNQX (3 μg/0.5 μl) infused into the NAC blocked the increase in locomotor activity produced by a dopamine agonist (David et al, 2004).
Lesion Procedure
Rats (Sprague–Dawley, derived from Charles River, but purchased from Harlan Laboratories SD) were anesthetized with chloral hydrate (400 mg/kg, i.p.) and placed in a Kopf stereotaxic instrument. The skin was retracted and holes were drilled in the skull above the NAC. Lesions were made by passing anodal current (0.5 mA for 55 s) through electrodes (Kopf Model NE-300 electrodes (0.25 mm in diameter and insulated to within 0.5 mm of the tip)) at the following co-ordinates AP=+1.2, ML=±1.2, DV=−7.6 and AP=+1.2, ML=±1.6, DV=−7.8 relative to bregma. Sham rats received that same treatment except that no current was passed through the electrode.
Apparatus
Experiment 1 was conducted at Yale University (apparatus as previously described by Cassella and Davis, 1986; Falls and Davis, 1997), while Experiment 2 was performed at the University of Northern Illinois (apparatus as previously described by Falls et al, 1997). In general, the apparatus used at both institutions had only minor differences (see below). Rats were trained and tested in stabilimeter devices. Each stabilimeter consisted of Plexiglas and wire-mesh cages (8 × 15 × 15 cm) suspended between compression springs within a steel frame housed in a sound-attenuated chamber (Industrial Acoustics Co., Model #105278, Bronx, NY). The sound-attenuated chamber was ventilated by a fan that also provided the background noise (55 dB sound pressure level (SPL A)). The floor of each stabilimeter consisted of four stainless steel bars through which a scrambled foot shock (0.5 s, 0.6 mA) could be delivered (generated by Lehigh Valley constant current shock generators, Model SGS-004, BRS/LVE, Beltsville, MD for Experiment 1 or Lafayette Instruments, Layfayette, IN, for Experiment 2). The startle-eliciting stimulus (a 105-dB, 50-ms burst of white noise with a rise/decay time of 4 ms) was provided by a white noise generator (Grasson-Stadler, Model 901B, West Concord, MA) connected to a 16.5 cm speaker (Alpine electronics, Model 6267 AX, Torrance, CA for Experiment 1, and Radio Shack Super Tweeter, Tandy Inc., Froth Worth, TX for Experiment 2) located behind each stabilimeter. The light CS was produced by an 8 W fluorescent light bulb (rise-decay time of 100 μs, 630 fL intensity) and controlled by a light control unit (Fintronics, Orange, CT). The noise CS (a 4-KHz, 70-dB band pass-filtered noise; 24 dB SPL per octave attenuation) was delivered through a full-range speaker (Radio Shack, #40-1286C) located 10 cm from the stabilimeter. In the Light-shock pairing condition of Experiment 1, the shock was presented 3.2 s after the onset of the 3.7-s light and in the Noise & Light-no shock condition, the 3.7-s noise was followed immediately by the 3.7-s light. During the test, the startle stimulus was presented 3.2 s after the onset of the light (eg when the shock previously occurred). For Experiment 2, the durations of the light CS and noise CS were 4 s. In the Light-shock pairing condition, the shock was presented 3.5 s after the onset of the light. During the test, the startle stimulus was presented 3.5 s after the onset of the light (eg when the shock previously occurred). Cage movement displaced an accelerometer (PCB Piezotronics, #321A, Depew, NY), which created a voltage change proportional to the velocity of the displacement. The accelerometer output was amplified (Fintronics Accelerometer, #FA 560, Orange, CT) and digitized (MacADIOS II Board, GW Instruments, Somerville, MA) on a 0-4096 unit scale. Startle amplitude was defined as the peak-to-peak accelerometer voltage that occurred during a 200-ms period after the onset of the startle stimulus. Data acquisition and stimulus presentations were controlled by a computer (Macintosh Power PC 7100/66).
Conditioned Inhibition of Fear-Potentiated Startle
Habituation
On each of 2 days before training, rats were placed in the startle chamber and 5 min later presented with 10 startle stimuli at each of 95, 100 and 105 dB intensities. The three intensities of startle stimuli were presented in a random order with an intertrial interval of 30 s.
Training
Training was conducted over two phases. During the first phase, the Light was paired with shock and during the second phase, the Noise & Light was presented with no shock. Phase 1 of training took place over 2 consecutive days during which animals were placed in the startle chamber and 5 min later received 10 Light-shock pairings with an overall intertrial interval of 2 min (range from 1–3 min). Phase 2 of training took place over 5 consecutive days during which rats were presented with five Light-shock trials intermixed with 15 Noise & Light-no shock trials. In these nonreinforced trials, the Noise CS was followed immediately by the Light and the shock was not presented. The two trial types (Light-shock and Noise & Light-no shock) were presented in a random sequence and the mean intertrial interval was 2 min (range between 1.5 and 2.5 min).
Test
To test for fear-potentiated startle and conditioned inhibition of fear-potentiated startle, rats were placed in the startle apparatus for 5 min and presented with 30 startle-eliciting stimuli alone (105 dB) followed by 15 startle stimuli (105 dB) in each of the three conditions: (1) in the dark with no explicit CS (embedded baseline startle trials), (2) in the presence of the Light (Light-startle trials to assess fear-potentiated startle), and (3) in the presence of the Noise & Light stimulus (Noise & Light-startle trial to assess conditioned inhibition of fear-potentiated startle). The order of the three trial types was randomized. All startle stimuli were presented with an interstimulus interval of 30 s.
Experiment 1
On tests days 1 and 2, rats were infused with amphetamine (0 and 20 μg/0.5 μl per side, in a counterbalanced order, n=15). On test day 3, a subset of rats (five chosen at random) was infused with 10 μg/0.5 μl of amphetamine. Rats were infused with CNQX (0 and 3 μg/0.5 μl per side, in a counterbalanced order, n=10) on test days 4 and 5. The tests were separated by at least 48 h (ie two training days).
Experiment 2
In Experiment 2a, rats received NAC lesion (n=9) or sham surgery (n=10) prior to training, while in Experiment 2b rats received post-training lesions of the NAC (n=14) or sham surgery (n=12) 24–48 h after the last day of training. Rats were allowed to recover for at least 7 days.
Prepulse Inhibition of Startle
The same apparatus as above was used to assess prepulse inhibition of the acoustic startle response (except that the background noise was 70 dB). Following a 5-min acclimation period, rats were presented with 10 100-dB startle stimuli, followed by 10 100-dB startle stimuli presented alone or 100 ms after a tone (2 kHz, 20 ms) at each of three intensities (72, 74, 78 dB). In all, 10 of each trial type (no prepulse (NP), 72, 74, and 78 dB prepulse) were presented in a random order. Animals were infused with amphetamine (0, 10 μg/0.5 μl) immediately prior to prepulse inhibition testing. A second test in which animals received the other dose of amphetamine was conducted 72 h later.
Histological Assessment
Upon completion of the experiment, rats were overdosed with chloral hydrate and perfused through the heart with PBS followed by 10% formaldehyde. Brains were sectioned (40 μm) and stained with cresyl violet to assess infusion site or lesion size and placement.
Statistical Analyses
An analysis of variance (ANOVA) was first conducted on the embedded baseline startle scores for each treatment group to determine if the treatment altered baseline startle responding. As there was no treatment effect on baseline startle scores in these experiments, these mean baseline startle scores were then subtracted from the mean Light-startle and mean Noise & Light-startle scores for each animal. The resulting difference scores reflect the magnitude of fear-potentiated startle (difference scores on Light-startle trials) and conditioned inhibition of fear-potentiated startle (difference scores on Noise & Light-startle trials). A second ANOVA, using Treatment (eg Lesion, Sham) as a between-subjects factor and Trial Type (Light, Noise & Light) as a within-subjects factor, was conducted on the difference scores to determine if the treatment affected fear-potentiated startle or conditioned inhibition of fear-potentiated startle.
Results
Experiment 1a: Effects of Amphetamine Infusions into the NAC on Fear-Potentiated Startle and Conditioned Inhibition of Fear-Potentiated Startle
Histology
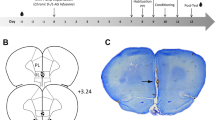
Figure 1a is a schematic representation of the infusion sites. As can be seen from this figure, all cannulae were placed within the boundaries of the NAC. Coronal sections are based on atlas plates from Paxinos and Watson (1998). Coordinates are in millimiter and relative to bregma.

Effects of increasing dopaminergic function or decreasing glutamatergic function on the acoustic startle modulation. (a) Drawings depicting the location of the microinjection cannula tips. Coronal sections are based on atlas plates from Paxinos and Watson (1998). Coordinates are in millimiter and relative to bregma. (b) Effects of intra-NAC infusions of amphetamine (0, 10 μg) on the expression of fear-potentiated startle and conditioned inhibition of fear-potentiated startle. The data shown are the mean (±SEM) difference scores computed by subtracting the mean baseline startle amplitude in startle alone test trials from the mean startle amplitude on Light trials (white bars) and Noise & Light trials (black bars). (c) Effects of intra-NAC infusions of CNQX (0, 3 μg) on the expression of fear-potentiated startle and conditioned inhibition of fear-potentiated startle. (d) Effects of intra-NAC infusions of amphetamine (0, 10 μg) on the expression of prepulse inhibition of startle. The data shown are the mean (±SEM) percent prepulse computed as ((amplitudes on prepulse trials/startle amplitudes on no prepulse trials) × 100%).
Intra-NAC amphetamine: baseline startle
The mean (±SEM) baseline (embedded) startle amplitudes for rats receiving amphetamine (0, 10, 20 μg) into the NAC were 248.32 (30.97), 280.37 (50.18), and 268.95 (38.64), respectively. An ANOVA performed on baseline startle amplitudes for animals that received two doses of amphetamine (0, 20 μg) showed no effect of amphetamine on baseline startle amplitudes (F(1,14)=0.37, p>0.05). An additional ANOVA performed on the data from the animals that received all doses of amphetamine (0, 10, and 20 μg) similarly showed no effect of amphetamine (F(2,8)=1.52, p>0.05). Thus, infusion of amphetamine into the NAC did not significantly affect baseline startle responding.
Intra-NAC amphetamine: fear-potentiated startle and conditioned inhibition of fear-potentiated startle
Figure 1b shows the effect of intra-NAC amphetamine on fear-potentiated startle (Light trials) and conditioned inhibition of fear-potentiated startle (Noise & Light trials). All groups show robust fear-potentiated startle and conditioned inhibition of fear-potentiated startle. The results of a mixed ANOVA performed on the data from animals receiving 0 and 20 μg of amphetamine support this interpretation showing a significant effect of Trial Type (F(1,14)=23.18, p<0.001), but no significant effect of Drug (F(1,14)=1.11, p>0.05) or interaction involving Drug (F(1,14)=1.95, p>0.05). An additional ANOVA performed on the data from the subgroup of animals that received all Drug treatments (0, 10, and 20 μg amphetamine) similarly showed only a significant effect of Trial type (F(1,4)=9.39, p<0.05) but no effect of Drug (F(2,8)=0.15, p>0.05) or interaction of Drug × Trial Type (F(2,8)=0.14, p>0.05). Thus, neither the expression of fear-potentiated startle or conditioned inhibition of fear-potentiated startle was affected by intra-NAC infusion of amphetamine.
Experiment 1b: Effects of CNQX Infusions into the NAC on Fear-Potentiated Startle and Conditioned Inhibition of Fear-Potentiated Startle
Intra-NAC CNQX: baseline startle
The mean (±SEM) baseline startle scores for rats receiving intra-NAC infusions of CNQX or vehicle were 419.47 (65.99) and 528.97 (77.0), respectively. An ANOVA performed on these scores showed no effect of the AMPA receptor antagonist on baseline startle amplitudes (F(1,9)=2.72, p>0.05). Thus, infusion of CNQX into the NAC did not significantly affect baseline startle responding.
Intra-NAC CNQX: fear-potentiated startle and conditioned inhibition of fear-potentiated startle
Figure 1c shows the effects of intra-NAC infusion of CNQX on fear-potentiated startle (Light trials) and conditioned inhibition of fear-potentiated startle (Noise & Light trials). Both vehicle and CNQX groups showed robust fear-potentiated startle and conditioned inhibition of fear-potentiated startle. An ANOVA showed a significant effect of Trial Type (F(1,9)=36.3, p<0.001), but no significant effect of Drug (F(1,9)=3.26, p>0.05) or interaction involving Drug (F(1,9)=1.18, p>0.05). Thus, fear-potentiated startle and conditioned inhibition of fear-potentiated startle are not affected by intra-NAC infusions of CNQX, showing that the expression of these processes does not depend on AMPA/kainate glutamate receptors in the NAC.
Experiment 1c: Effects of Amphetamine Infusions into the NAC on Prepulse Inhibition of the Acoustic Startle Response
The mean (±SEM) startle amplitude on baseline and prepulse (NP, 72, 74, and 78 dB) trials following infusion of amphetamine (0 or 10 μg) were 625.92 (112.16), 338.52 (45.52), 322.89 (53.19), and 237.30 (38.49) for vehicle and 520.20 (110.58), 469.02 (102.87), 442.46 (82.44), and 260.82 (59.63), for amphetamine, respectively.
For the sake of clarity, the prepulse inhibition scores are presented as percent prepulse inhibition ((startle amplitude on prepulse trials/startle amplitude on baseline trials) × 100%) in Figure 1d. Prepulse inhibition using prepulses of lower intensity was significantly disrupted by intra-NAC infusions of amphetamine. An ANOVA using Drug (amphetamine vs vehicle) as a between-subjects factor and Trial Type (NP, 72, 74, 78 dB prepulse intensity) as a within-subjects factors shows a significant Drug × Trial Type interaction (F(3,27)=12.45, p<0.001) and significant effects of Trial Type (F(3,27)=65.24, p<0.001) and Drug (F(1,9)=26.59, p<0.001). Post hoc Newman–Keul comparisons show that the percentage of prepulse inhibition on all prepulse trials (72, 74, and 78 dB) was significantly lower (p<0.05) than on startle alone (no prepulse trials) for vehicle-infused rats. However, in amphetamine-treated rats, prepulse inhibition was significantly different from startle alone trials following only the most intense prepulse stimulus (78 dB). Thus, in accordance with previous studies, intra-NAC infusions of amphetamine disrupt prepulse inhibition of the acoustic startle response. It is important to point out, however, that this dose of amphetamine, in the same anatomical region in the same rats, had no effect on fear-potentiated startle or conditioned inhibition of fear-potentiated startle.
Experiment 2a: Effect of Pretraining NAC Lesions on the Acquisition of Fear-Potentiated Startle and Conditioned Inhibition of Fear-Potentiated Startle
Pre-training electrolytic lesion of the NAC: lesion assessment
Figure 2a shows a schematic representation of the damage produced by the electrolytic lesions. Representative boundaries of the largest (gray) and the smallest (black) lesions of the NAC are depicted. Lesioned rats sustained extensive bilateral damage to the NAC, involving both the core and shells regions over the entire rostral-caudal extent of the NAC. These lesions produced limited damage to adjacent regions, including the ventral pallidum, lateral septal nucleus, and the medial forebrain bundle. Sham rats showed a small amount of cortical damage caused by lowering the electrode but there was no evidence of damage to the NAC.

Effects of pretraining (b) and post-training (c) electrolytic lesions of the NAC on the acquisition and expression of fear-potentiated startle and conditioned inhibition of fear-potentiated startle. (a) Representation of the minimum (black) and maximum (gray) damage produced by electrolytic lesions to the NAC in Experiment 1. The coronal section is based on atlas plates from Paxinos and Watson (1998). Coordinates are in millimiter and relative to bregma. (b) Effects of pretraining electrolytic lesions of the NAC on the acquisition of fear potentiated startle and conditioned inhibition of fear-potentiated startle. The data shown are the mean (±SEM) difference scores computed by subtracting the mean baseline startle amplitude in startle alone test trials from the mean startle amplitude on Light trials (white bars) and Noise & Light trials (black bars). (c) Effects of post-training electrolytic lesions of the NAC on the expression of fear-potentiated startle and conditioned inhibition of fear-potentiated startle.
Pre-training electrolytic lesion of NAC: baseline startle
The mean (±SEM) baseline startle scores for Lesion (872.81±163.21) and Sham (1000.55±148.10) treatment groups did not differ (F(1,17)=0.34, p>0.05). Thus, pretraining electrolytic lesions of the NAC do not significantly affect baseline startle amplitude.
Pretraining electrolytic lesion of NAC: fear-potentiated startle and conditioned inhibition of fear-potentiated startle
Figure 2b shows the effects of pretraining lesions of the NAC on the acquisition of fear-potentiated startle (Light trials) and conditioned inhibition of fear-potentiated startle (Noise & Light trials). Both Lesion and Sham treatment groups show high levels of fear-potentiated startle and substantially lower responding on conditioned inhibition trials. The results of a mixed ANOVA support this interpretation, showing a significant effect of Trial Type (F(1,17)=16.57, p<0.001) but no significant effect of Lesion (F(1,17)=0.31, p>0.05) or interaction between Trial Type and Lesion (F(1,17)=0.28, p>0.05). Therefore, pretraining electrolytic lesions of the NAC had no effect on the acquisition of fear-potentiated startle or conditioned inhibition of fear-potentiated startle.
Experiment 2b: Effect of Post-Training NAC Lesions on the Expression of Fear-Potentiated Startle and Conditioned Inhibition of Fear-Potentiated Startle
Post-training electrolytic lesion of the NAC: lesion assessment
Similar to above, electrolytic lesions of the NAC produced substantial bilateral damage to the core and shell regions of the NAC. The sham control animals showed no damage to the NAC.
Post-training electrolytic lesion of the NAC: baseline startle
The mean (±SEM) baseline startle scores for Lesioned (648.56±54.1) and Sham (586.22±84.95) animals did not differ (F(1,24)=0.42, p>0.05). Therefore, NAC lesions performed either before (above) or after (present) training do not significantly affect the amplitude of baseline startle responses.
Post-training electrolytic lesion of the NAC: fear-potentiated startle and conditioned inhibition of fear-potentiated startle
Figure 2c shows the effects of post-training NAC lesions on the performance of fear-potentiated startle (Light trials) and conditioned inhibition of fear-potentiated startle (Noise & Light trials). Both Lesion and Sham treatment groups show high responding on the Light trials, indicating robust fear-potentiated startle, and substantially lower responding on the Noise & Light trials, indicated conditioned inhibition of fear-potentiated startle. The results of a mixed ANOVA support this interpretation, showing a significant effect of Trial Type (F(1,24)=32.02, p<0.001), but no significant effect of Lesion (F(1,24)=1.87, p>0.05) or interaction between Trial Type and Lesion (F(1,24)=1.48, p>0.05). Therefore, lesions of the NAC do not affect the retrieval or performance of fear-potentiated startle or conditioned inhibition of fear-potentiated startle.
Discussion
The acoustic startle reflex is sensitive to the emotional and/or motivational state of the subject. The startle reflex in humans is increased in the presence of cues signaling danger and decreased if elicited in a pleasant or rewarding emotional context (Vrana et al, 1988; Grillon et al, 1993). Likewise rodents show increased startle in the presence of cues paired with shock (Davis, 1992) and drugs that increase anxiety (Frankland et al, 1997) and decreased startle in the presence of cues paired with food (Schmid et al, 1995) or rewarding brain stimulation (Steidl et al, 2001).
The present series of experiments took advantage of the affective modulation of the acoustic startle response to investigate the role of the NAC in mediating cues signaling danger (shock) and safety (no shock). In this study, we paired a Light stimulus with shock and a Noise & Light stimulus with no shock. Previous experiments using this procedure (Falls and Davis, 1997) show that the Noise inhibits conditioned fear through Pavlovian conditioned inhibition rather than through other nonassociative mechanisms such as generalization decrement or ‘external’ inhibition (Pavlov, 1927). One advantage of this type of conditioned inhibition procedure is that the effects of NAC manipulations on fear excitation and inhibition can be assessed simultaneously.
The present study examined the role of the NAC in mediating the behavioral effect of a safety signal using acute drug manipulations and permanent lesions of the NAC. As previous research suggests that dopamine- and glutamate-mediated mechanisms are key to NAC function (Choi et al, 2000; David et al, 2004), we examined the effects of disrupting AMPA/kainate receptor function and potentiating dopamine function in the NAC. Neither manipulation altered the expression of fear-potentiated startle or conditioned inhibition of fear-potentiated startle. However, intra-NAC infusion of amphetamine did disrupt prepulse inhibition of startle in the same animals. These positive control data are consistent with previous reports (Swerdlow et al, 1986; Wan et al, 1995) and show that our treatment produced adequate perfusion of the NAC. In addition, large pre- or post-training lesions of the NAC had no impact on the acquisition or retention of fear-potentiated startle or conditioned inhibition of fear-potentiated startle. Taken together, the present results clearly show that the NAC is not critical for the acquisition or expression of fear-potentiated startle or conditioned inhibition of fear-potentiated startle.
These negative findings are surprising for two reasons. First, some learning theories postulate the existence of two mutually antagonistic motivational systems in the brain: the appetitive motivational system (that directs behavior towards an attractive stimulus such as food) and aversive motivational system (that directs behavior away from an aversive stimulus such as shock) (Konorski, 1967; Dickinson and Dearing, 1979). As these motivational systems would reciprocally inhibit one another, activation of the aversive system would inhibit the appetitive motivational system. By extension, a safety signal that inhibits the aversive system may be perceived by the animal as motivationally equivalent to a CS that activates the appetitive motivational system. Indeed, there is experimental evidence supporting this opponent process model of appetitive-aversive interactions (Denny, 1971; Dinsmoor, 2001; Pineno, 2004). For instance, the omission of food (an appetitive US) and a CS associated with such an omission shows aversive properties (Amsel, 1958; Leitenberg, 1965; Coughlin, 1972). On the other hand, a CS associated with the omission of an expected shock (a safety signal) shows appetitive properties such as the ability to support lever pressing (Hendry, 1967; DeVito and Fowler, 1986, 1994; Fowler et al, 1977). It follows, therefore, that the neural structures that mediate the behavioral effects of safety signals may overlap with those that mediate appetitive conditioning. As previous data shows that the NAC is critically involved in mediating conditioned appetitive states (Balleine and Killcross, 1994; Parkinson et al, 2000; Corbit et al, 2001; Hall et al, 2001), it is surprising that similar manipulations of NAC function did not affect conditioned inhibition of fear. It should be pointed out, however, that we did not directly assess whether our safety signal manipulation produced an appetitive state.
Second, the NAC has been implicated in mediating the effects of an appetitive Pavlovian cue on startle (Koch et al, 2000). In this ‘reward-attenuated startle’ paradigm, presentation of a CS previously paired with food availability reduced baseline startle responding (Schmid et al, 1995; Koch et al, 2000). Importantly, pretraining 6-hydroxydopamine lesions of the NAC block this effect (Koch et al, 1996). Therefore, the NAC is important for the inhibition of startle by a cue signaling food availability but not by a cue signaling safety, indicating that the neural circuits mediating these processes are dissociable. In this sense, brain areas responsible for processing a safety signal are not identical to those responsible for processing appetitive Pavlovian cues. The present results, therefore, indicate important neural dissociations between the processing of an appetitive signal and a safety signal.
The NAC has also been implicated in another type of inhibitory learning, latent-inhibition. Latent inhibition is a phenomenon in which repeated exposure to a stimulus without reinforcement retards subsequent conditioning to that stimulus (Lubow, 1973; Lubow and Gewirtz, 1995). Importantly, lesions of the shell region of the NAC attenuate the effect of stimulus pre-exposure (the latent inhibition effect) in a fear conditioning paradigm (Weiner et al, 1996; Murphy et al, 2000). Therefore, while both latent inhibition and conditioned inhibition of fear reduce conditioned fear responses, these two processes depend on different anatomical systems.
In addition to the finding that the NAC is not critically involved in conditioned inhibition of fear-potentiated startle, our study examined the role of the NAC in fear-potentiated startle. Previous findings on the role of the NAC in fear conditioning are mixed. While some studies show that stimuli previously paired with shock are associated with an increase NAC DA release (Wilkinson et al, 1998; Young et al, 1998; Pezze et al, 2001) and the expression of plasticity-associated immediate early genes (Beck and Fibiger, 1995; Thomas et al, 2002), other studies have failed to find change in DA release (Levita et al, 2002). These discrepancies may be due to procedural differences and the heterogeneity of the NAC. The present findings show that manipulations of the NAC (including large electrolytic lesions) do not affect acquisition or retention of conditioned fear as measured by fear-potentiated startle. Our results support growing evidence that lesions (Riedel et al, 1997; Levita et al, 2002; Jongen-Relo et al, 2003), temporary inactivation (Haralambous and Westbrook, 1999), or morphine infusions into the NAC (Westbrook et al, 1997) do not disrupt conditioned fear to discrete cues, such as tones or lights, regardless of how conditioned fear is measured (fear-potentiated startle or conditioned freezing). The NAC has been implicated, however, in contextual fear conditioning (without a discrete cue such as a light or a tone) (Westbrook et al, 1997; Haralambous and Westbrook, 1999; Levita et al, 2002), perhaps through hippocampal efferents via the ventral subiculum (Groenewegen et al, 1987). It has been suggested that the locomotor exploration required for processing contextual stimuli may be mediated by interactions between the NAC and hippocampus (Maren et al, 1997; Fanselow, 2000; Thomas et al, 2002).
The neural substrates that mediate safety signals are unclear. Although the amygdala has been shown to be critically involved in the acquisition and expression of conditioned fear, lesions of the central nucleus of the amygdala do not disrupt the effects of a safety signal on conditioned fear (Falls and Davis, 1995). While picrotoxin injections into the dorsal periaqueductal gray decrease the expression of conditioned inhibition of fear-potentiated startle (Fendt, 1998), lesions of prefrontal cortex (Gewirtz et al, 1997), perirhinal cortex (Falls et al, 1997), and auditory thalamus (Heldt and Falls, 1998) fail to disrupt it. Lesions of the hippocampus performed after training specifically disrupted the effects of the safety signal on conditioned fear responses without disrupting conditioned fear responses themselves (Heldt et al, 2002). However, the findings that further training overcomes the effect of the lesions and that similar lesions performed before training failed to affect the impact of the safety signal suggests that additional brain regions are also critically involved. Although the hippocampal–NAC axis has been emphasized in the flow of information (Grace, 2000), the present findings indicate that the NAC is not critically involved in conditioned inhibition of fear-potentiated startle.
A reduced ability to inhibit conditioned fear associations or responses when the threat is no longer relevant might contribute to the persistence of maladaptive fear. Indeed, failure of these inhibitory mechanisms may lead to clinical conditions such as pathological anxiety (Rosen and Schulkin, 1998; Bouton et al, 2001; Myers and Davis, 2002). Therefore, a greater understanding of the brain regions involved in the inhibition of fear, and the relationships between these regions, will enhance our understanding of fear processes and have important clinical implications for the treatment of disorders that associated with dysfunction of the inhibition of fear such as anxiety disorders.
References
Amsel A (1958). The role of frustrative nonreward in noncontinous reward situations. Psychol Bull 55: 102–119.
Balleine B, Killcross S (1994). Effects of ibotenic acid lesions of the nucleus accumbens on instrumental action. Behav Brain Res 65: 181–193.
Beck CH, Fibiger HC (1995). Conditioned fear-induced changes in behavior and in the expression of the immediate early gene c-fos: with and without diazepam pretreatment. J Neurosci 15: 709–720.
Berridge KC, Robinson TE (1998). What is the role of dopamine in reward: hedonic impact, reward learning, or incentive salience? Brain Res Brain Res Rev 28: 309–369.
Bouton ME, Mineka S, Barlow DH (2001). A modern learning theory perspective on the etiology of panic disorder. Psychol Rev 108: 4–32.
Brog JS, Salyapongse A, Deutch AY, Zahm DS (1993). The patterns of afferent innervation of the core and shell in the ‘accumbens’ part of the rat ventral striatum: immunohistochemical detection of retrogradely transported fluoro-gold. J Comp Neurol 338: 255–278.
Brown PL, Jenkins HM (1968). Auto-shaping of the pigeon's key-peck. J Exp Anal Behav 11: 1–8.
Cain CK, Blouin AM, Barad M (2003). Temporally massed CS presentations generate more fear extinction than spaced presentations. J Exp Psychol Anim Behav Process 29: 323–333.
Campeau S, Falls WA, Cullinan WE, Helmreich DL, Davis M, Watson SJ (1997). Elicitation and reduction of fear: behavioural and neuroendocrine indices and brain induction of the immediate-early gene c-fos. Neuroscience 78: 1087–1104.
Cassella JV, Davis M (1986). The design and calibration of a startle measurement system. Physiol Behav 36: 377–383.
Choi KH, Zarandi B, Todd KG, Biondo AM, Greenshaw AJ (2000). Effects of AMPA/kainate receptor blockade on responses to dopamine receptor agonists in the core and shell of the rat nucleus accumbens. Psychopharmacology (Berl) 150: 102–111.
Corbit LH, Muir JL, Balleine BW (2001). The role of the nucleus accumbens in instrumental conditioning: evidence of a functional dissociation between accumbens core and shell. J Neurosci 21: 3251–3260.
Coughlin RC (1972). The aversive properties of withdrawing positive reinforcement: a review of the recent literature. Psychological Record 22: 333–354.
David HN, Sissaoui K, Abraini JH (2004). Modulation of the locomotor responses induced by D(1)-like and D(2)-like dopamine receptor agonists and D-amphetamine by NMDA and non-NMDA glutamate receptor agonists and antagonists in the core of the rat nucleus accumbens. Neuropharmacology 46: 179–191.
Davis M (1992). The role of the amygdala in fear and anxiety. Annu Rev Neurosci 15: 353–375.
Dayan P, Balleine BW (2002). Reward, motivation, and reinforcement learning. Neuron 36: 285–298.
de Borchgrave R, Rawlins JN, Dickinson A, Balleine BW (2002). Effects of cytotoxic nucleus accumbens lesions on instrumental conditioning in rats. Exp Brain Res 144: 50–68.
Denny MR (1971). Relaxation theory and experiments. In: Brush FR (ed). Aversive Conditioning and Learning. Academic Press: New York, NY.
DeVito PL, Fowler H (1986). Effects of contingency violations on the extinction of a conditioned fear inhibitor and a conditioned fear excitor. J Exp Psychol Anim Behav Process 12: 99–115.
DeVito PL, Fowler H (1994). Positive and negative transfer of conditioned aversive stimuli to a conditioned appetitive excitor as a function of aversive US intensity. Animal Learn Behav 22: 195–202.
Dickinson A, Dearing MF (1979). Appetitive–aversive interactions and inhibitory processes. In: Dickinson A, Boakes A (eds). Mechanisms of Learning and Motivation: a Memorial Volume to Jerzy Konorksi. Erlbaum: Hillsdale, NJ. pp 203–231.
Dinsmoor JA (2001). Stimuli inevitably generated by behavior that avoids electric shock are inherently reinforcing. J Exp Anal Behav 75: 311–333.
Everitt BJ, Wolf ME (2002). Psychomotor stimulant addiction: a neural systems perspective. J Neurosci 22: 3312–3320.
Falls WA, Bakken KT, Heldt SA (1997). Lesions of the perirhinal cortex interfere with conditioned excitation but not with conditioned inhibition of fear. Behav Neurosci 111: 476–486.
Falls WA, Davis M (1995). Lesions of the central nucleus of the amygdala block conditioned excitation, but not conditioned inhibition of fear as measured with the fear-potentiated startle effect. Behav Neurosci 109: 379–387.
Falls WA, Davis M (1997). Inhibition of fear-potentiated startle can be detected after the offset of a feature trained in a serial feature-negative discrimination. J Exp Psychol Anim Behav Process 23: 3–14.
Fanselow MS (2000). Contextual fear, gestalt memories, and the hippocampus. Behav Brain Res 110: 73–81.
Fanselow MS, Gale GD (2003). The amygdala, fear, and memory. Ann NY Acad Sci 985: 125–134.
Fendt M (1998). Different regions of the periaqueductal grey are involved differently in the expression and conditioned inhibition of fear-potentiated startle. Eur J Neurosci 10: 3876–3884.
Fowler H, Goodman JH, DeVito PL (1977). Across-reinforcement blocking effects in a mediational test of the CS's general signalling property. Learn Motivat 8: 507–519.
Frankland PW, Josselyn SA, Bradwejn J, Vaccarino FJ, Yeomans JS (1997). Activation of amygdala cholecystokininB receptors potentiates the acoustic startle response in the rat. J Neurosci 17: 1838–1847.
Gale GD, Anagnostaras SG, Godsil BP, Mitchell S, Nozawa T, Sage JR et al (2004). Role of the basolateral amygdala in the storage of fear memories across the adult lifetime of rats. J Neurosci 24: 3810–3815.
Gewirtz JC, Falls WA, Davis M (1997). Normal conditioned inhibition and extinction of freezing and fear-potentiated startle following electrolytic lesions of medical prefrontal cortex in rats. Behav Neurosci 111: 712–726.
Grace AA (2000). Gating of information flow within the limbic system and the pathophysiology of schizophrenia. Brain Res Brain Res Rev 31: 330–341.
Grillon C, Ameli R, Foot M, Davis M (1993). Fear-potentiated startle: relationship to the level of state/trait anxiety in healthy subjects. Biol Psychiatry 33: 566–574.
Groenewegen HJ, Vermeulen-Van der Zee E, te Kortschot A, Witter MP (1987). Organization of the projections from the subiculum to the ventral striatum in the rat. A study using anterograde transport of Phaseolus vulgaris leucoagglutinin. Neuroscience 23: 103–120.
Groenewegen HJ, Wright CI, Beijer AV, Voorn P (1999). Convergence and segregation of ventral striatal inputs and outputs. Ann NY Acad Sci 877: 49–63.
Hall J, Parkinson JA, Connor TM, Dickinson A, Everitt BJ (2001). Involvement of the central nucleus of the amygdala and nucleus accumbens core in mediating Pavlovian influences on instrumental behaviour. Eur J Neurosci 13: 1984–1992.
Haralambous T, Westbrook RF (1999). An infusion of bupivacaine into the nucleus accumbens disrupts the acquisition but not the expression of contextual fear conditioning. Behav Neurosci 113: 925–940.
Heldt SA, Coover GD, Falls WA (2002). Posttraining but not pretraining lesions of the hippocampus interfere with feature-negative discrimination of fear-potentiated startle. Hippocampus 12: 774–786.
Heldt SA, Falls WA (1998). Destruction of the auditory thalamus disrupts the production of fear but not the inhibition of fear conditioned to an auditory stimulus. Brain Res 813: 274–282.
Hendry DP (1967). Conditioned inhibition of conditioned suppression. Psychonomic Soc 9: 261–262.
Jongen-Relo AL, Kaufmann S, Feldon J (2003). A differential involvement of the shell and core subterritories of the nucleus accumbens of rats in memory processes. Behav Neurosci 117: 150–168.
Koch M, Schmid A, Schnitzler HU (1996). Pleasure-attenuation of startle is disrupted by lesions of the nucleus accumbens. Neuroreport 7: 1442–1446.
Koch M, Schmid A, Schnitzler HU (2000). Role of muscles accumbens dopamine D1 and D2 receptors in instrumental and Pavlovian paradigms of conditioned reward. Psychopharmacology (Berl) 152: 67–73.
Konorski J (1967). Integrative Activity of the Brain. University of Chicago Press: Chicago.
LeDoux JE (2000). Emotion circuits in the brain. Annu Rev Neurosci 23: 155–184.
Leitenberg M (1965). Is time out from positive reinforcement an aversive event? A review of the experimental evidence. Psychol Bull 64: 428–441.
Levita L, Dalley JW, Robbins TW (2002). Nucleus accumbens dopamine and learned fear revisited: a review and some new findings. Behav Brain Res 137: 115–127.
Lubow RE (1973). Latent inhibition. Psychol Bull 79: 398–407.
Lubow RE, Gewirtz JC (1995). Latent inhibition in humans: data, theory and implications for schizophrenia. Psychol Bull 117: 87–103.
Maren S (2003). The amygdala, synaptic plasticity, and fear memory. Ann NY Acad Sci 985: 106–113.
Maren S, Aharonov G, Fanselow MS (1997). Neurotoxic lesions of the dorsal hippocampus and Pavlovian fear conditioning in rats. Behav Brain Res 88: 261–274.
McDonald AJ (1991). Topographical organization of amygdaloid projections to the caudatoputamen, nucleus accumbens, and related striatal-like areas of the rat brain. Neuroscience 44: 15–33.
Murphy CA, Peeze M-A, Feldon J, Heidbreder C (2000). Differential involvement of dopamine in the shell and core of the nucleus accumbens in the expression of latent inhibition to an aversively conditioned stimulus. Neuroscience 97: 469–477.
Myers KM, Davis M (2002). Behavioral and neural analysis of extinction. Neuron 36: 567–584.
Parkinson JA, Olmstead MC, Burns LH, Robbins TW, Everitt BJ (1999). Dissociation in effects of lesions of the nucleus accumbens core and shell on appetitive pavlovian approach behavior and the potentiation of conditioned reinforcement and locomotor activity by D-amphetamine. J Neurosci 19: 2401–2411.
Parkinson JA, Willoughby PJ, Robbins TW, Everitt BJ (2000). Disconnection of the anterior cingulate cortex and nucleus accumbens core impairs Pavlovian approach behavior: further evidence for limbic cortical-ventral striatopallidal systems. Behav Neurosci 114: 42–63.
Pavlov I (1927). Conditioned Reflexes. Oxford University Press: Oxford.
Paxinos G, Watson C (1998). The Rat Brain in Stereotaxoc Co-prdoamtes. Academic Press: NewYork.
Pezze MA, Heidbreder CA, Feldon J, Murphy CA (2001). Selective responding of nucleus accumbens core and shell dopamine to aversively conditioned contextual and discrete stimuli. Neuroscience 108: 91–102.
Pineno O (2004). Differential effects of nonreinforcement and punishment in humans. Psycologica 25: 87–102.
Quirk GJ, Gehlert DR (2003). Inhibition of the amygdala: key to pathological states? Ann NY Acad Sci 985: 263–272.
Rescorla RA (1969). Pavlovian conditioned inhibition. Psychol Bull 72: 77–94.
Riedel G, Harrington NR, Hall G, Macphail EM (1997). Nucleus accumbens lesions impair context, but not cue, conditioning in rats. Neuroreport 8: 2477–2481.
Robbins TW, Cador M, Taylor JR, Everitt BJ (1989). Limbic–striatal interactions in reward-related processes. Neurosci Biobehav Rev 13: 155–162.
Rogan MT, Leon KS, Perez DL, Kandel ER (2003). Danger and safety learning in the mouse: amygdala involvement in negative and positive affective responses Society for Neurosciences Abstracts Program 624.9.
Rosen JB, Schulkin J (1998). From normal fear to pathological anxiety. Psychol Rev 105: 325–350.
Schafe GE, Nader K, Blair HT, LeDoux JE (2001). Memory consolidation of Pavlovian fear conditioning: a cellular and molecular perspective. Trends Neurosci 24: 540–546.
Schmid A, Koch M, Schnitzler HU (1995). Conditioned pleasure attenuates the startle response in rats. Neurobiol Learn Mem 64: 1–3.
Steidl S, Li L, Yeomans JS (2001). Conditioned brain-stimulation reward attenuates the acoustic startle reflex in rats. Behav Neurosci 115: 710–717.
Swerdlow NR, Braff DL, Geyer MA, Koob GF (1986). Central dopamine hyperactivity in rats mimics abnormal acoustic startle response in schizophrenics. Biol Psychiatry 21: 23–33.
Thomas KL, Hall J, Everitt BJ (2002). Cellular imaging with zif268 expression in the rat nucleus accumbens and frontal cortex further dissociates the neural pathways activated following the retrieval of contextual and cued fear memory. Eur J Neurosci 16: 1789–1796.
Vrana SR, Spence EL, Lang PJ (1988). The startle probe response: a new measure of emotion? J Abnorm Psychol 97: 487–491.
Wallace KJ, Rosen JB (2001). Neurotoxic lesions of the lateral nucleus of the amygdala decrease conditioned fear but not unconditioned fear of a predator odor: comparison with electrolytic lesions. J Neurosci 21: 3619–3627.
Wan FJ, Geyer MA, Swerdlow NR (1995). Presynaptic dopamine–glutamate interactions in the nucleus accumbens regulate sensorimotor gating. Psychopharmacology (Berl) 120: 433–441.
Weiner I, Gal G, Rawlins JNP, Feldon J (1996). Differential involvement of the shell and core subterritories of the nucleus accumbens in latent inhibition and amphetamine-induded activity. Behav Brain Res 81: 123–133.
Westbrook RF, Good AJ, Kiernan MJ (1997). Microinjection of morphine into the nucleus accumbens impairs contextual learning in rats. Behav Neurosci 111: 996–1013.
Wilkinson LS, Humby T, Killcross AS, Torres EM, Everitt BJ, Robbins TW (1998). Dissociations in dopamine release in medial prefrontal cortex and ventral striatum during the acquisition and extinction of classical aversive conditioning in the rat. Eur J Neurosci 10: 1019–1026.
Wyvell CL, Berridge KC (2000). Intra-accumbens amphetamine increases the conditioned incentive salience of sucrose reward: enhancement of reward ‘wanting’ without enhanced ‘liking’ or response reinforcement. J Neurosci 20: 8122–8130.
Young AM, Ahier RG, Upton RL, Joseph MH, Gray JA (1998). Increased extracellular dopamine in the nucleus accumbens of the rat during associative learning of neutral stimuli. Neuroscience 83: 1175–1183.
Zahm DS (2000). An integrative neuroanatomical perspective on some subcortical substrates of adaptive responding with emphasis on the nucleus accumbens. Neurosci Biobehav Rev 24: 85–105.
Acknowledgements
This work was supported by a National Institute of Mental Health Grants MH-57250 and MH-47840, Research Scientist Development Award MH-0004, a grant from the Air Force Office of Scientific Research, and the State of Connecticut to MD and a National Alliance for Research on Schizophrenia and Depression (NARSAD) Young Investigator Award to SAJ. We thank Bernard Balleine and Paul W Frankland for helpful discussions and comments on an earlier version of this paper.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Josselyn, S., Falls, W., Gewirtz, J. et al. The Nucleus Accumbens is not Critically Involved in Mediating the Effects of a Safety Signal on Behavior. Neuropsychopharmacol 30, 17–26 (2005). https://doi.org/10.1038/sj.npp.1300530
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.npp.1300530
Keywords
This article is cited by
-
Meta-analysis of human prediction error for incentives, perception, cognition, and action
Neuropsychopharmacology (2022)
-
Chronic inhibition of GABA synthesis in the infralimbic cortex facilitates conditioned safety memory and reduces contextual fear
Translational Psychiatry (2020)
-
Infralimbic cortex activity is required for the expression but not the acquisition of conditioned safety
Psychopharmacology (2020)
-
Paradoxical Neurobehavioral Rescue by Memories of Early-Life Abuse: The Safety Signal Value of Odors Learned during Abusive Attachment
Neuropsychopharmacology (2015)
-
The Role of the Nucleus Accumbens Shell in the Mediation of the Reinforcing Properties of a Safety Signal in Free-Operant Avoidance: Dopamine-Dependent Inhibitory Effects of d-amphetamine
Neuropsychopharmacology (2014)
