
Abstract
Atypical antipsychotic drugs have revolutionized the treatment of schizophrenia and related disorders. The current clinically approved atypical antipsychotic drugs are characterized by having relatively low affinities for D2-dopamine receptors and relatively high affinities for 5-HT2A serotonin receptors (5-HT, 5-hydroxytryptamine (serotonin)). Aripiprazole (OPC-14597) is a novel atypical antipsychotic drug that is reported to be a high-affinity D2-dopamine receptor partial agonist. We now provide a comprehensive pharmacological profile of aripiprazole at a large number of cloned G protein-coupled receptors, transporters, and ion channels. These data reveal a number of interesting and potentially important molecular targets for which aripiprazole has affinity. Aripiprazole has highest affinity for h5-HT2B-, hD2L-, and hD3-dopamine receptors, but also has significant affinity (5–30 nM) for several other 5-HT receptors (5-HT1A, 5-HT2A, 5-HT7), as well as α1A-adrenergic and hH1-histamine receptors. Aripiprazole has less affinity (30–200 nM) for other G protein-coupled receptors, including the 5-HT1D, 5-HT2C, α1B-, α2A-, α2B-, α2C-, β1-, and β2-adrenergic, and H3-histamine receptors. Functionally, aripiprazole is an inverse agonist at 5-HT2B receptors and displays partial agonist actions at 5-HT2A, 5-HT2C, D3, and D4 receptors. Interestingly, we also discovered that the functional actions of aripiprazole at cloned human D2-dopamine receptors are cell-type selective, and that a range of actions (eg agonism, partial agonism, antagonism) at cloned D2-dopamine receptors are possible depending upon the cell type and function examined. This mixture of functional actions at D2-dopamine receptors is consistent with the hypothesis proposed by Lawler et al (1999) that aripiprazole has ‘functionally selective’ actions. Taken together, our results support the hypothesis that the unique actions of aripiprazole in humans are likely a combination of ‘functionally selective’ activation of D2 (and possibly D3)-dopamine receptors, coupled with important interactions with selected other biogenic amine receptors—particularly 5-HT receptor subtypes (5-HT1A, 5-HT2A).
Similar content being viewed by others

INTRODUCTION
Schizophrenia, a neuropsychiatric disorder affecting more than 1% of the world's population, is a devastating and costly disease. As of 1991, it was estimated that costs associated with schizophrenia in the United States approached $65 billion per year (Wyatt et al, 1995). Costs are high because schizophrenia is a chronic disease, it affects people in their youth, and treatment often requires frequent and intensive hospitalization and outpatient care. Schizophrenia is characterized by the appearance of delusions and hallucinations (positive symptoms), but may involve a variety of other symptoms including decreased social functioning and speech, lack of motivation, poor hygiene, and disorganization (all negative symptoms), as well as cognitive impairment.
Effective medication-based treatments for schizophrenia have been available for more than four decades. Early therapies were based on what are now called typical antipsychotics (eg chlorpromazine, haloperidol, and fluphenazine). These drugs were effective in reducing the positive symptoms in 70–80% of schizophrenics, but tended to cause acute extrapyramidal side effects (EPS) such as Parkinsonism and acute dystonias, a lack of improvement (or a worsening) of negative symptoms, elevation of serum prolactin levels, and (with chronic use) the debilitating neurological side effect tardive dyskinesia. A common feature of typical antipsychotic drugs is their affinity for D2-dopamine receptors, typically in the low nanomolar (<2.0 nM) range (Creese et al, 1976)
Clozapine, the prototypical atypical antipsychotic, was reintroduced in the late 1980s following a pivotal study demonstrating its effectiveness in treatment-resistant schizophrenia (Kane et al, 1988). Since then, other atypical antipsychotic drugs from several chemical classes, including dibenzodiazepines (clozapine and quetiapine), thienobenzodiazepines (olanzapine), benzisothiazolyl piperazines (ziprasidone), and benzisoxazoles (risperidone), have been introduced. Unlike the typical antipsychotics, it has been difficult to find a common mechanism explaining the actions of these drugs. With the exception of quetiapine, the atypicals have 5-HT2A/D2 affinity ratios grater than 10 (Meltzer et al, 1989), but they also interact with other receptors from the serotonin (5-HT1A, 5-HT2C, 5-HT6, 5-HT7) (Roth et al, 1992,1994,1998), dopamine (D3, D4) (Roth et al, 1995; Sokoloff et al, 1990; Van Tol et al, 1991), muscarinic cholinergic (Stanton et al, 1993; Zeng et al, 1997), and histamine families (Kroeze et al, 2003; Roth et al, in press). In fact, the useful properties of the atypicals may depend on simultaneous effects caused by interactions with multiple G protein-coupled receptors (GPCRs) (Roth, 1994,2000). From this perspective, it is not surprising that atypical antipsychotic drugs are effective in bipolar and schizoaffective disorders and depression (Ghaemi et al, 2000; Narendran et al, 2001; Sanger et al, 2001).
Recently, the FDA approved aripiprazole, a new atypical antipsychotic drug (Inoue et al, 1997; Inoue et al, 1996; Oshiro et al, 1998) that is proposed to differ in mechanism of action from other atypical antipsychotic drugs. Aripiprazole has high affinity for D2- and D3-dopamine and 5-HT7 serotonin receptors (Lawler et al, 1999). There has been a long-standing hypothesis that D2 partial agonists would be of particular utility in schizophrenia (Tamminga, 2002), and the developers of aripiprazole have proposed that the novel and improved clinical profile of aripiprazole is caused by the partial agonist properties at D2-dopamine receptors that result in ‘dopamine stabilization’ (Burris et al, 2002; Inoue et al, 1996). Conversely, it has been suggested (Lawler et al, 1999) that aripiprazole is not simply a partial agonist, but a drug whose D2 functional effects were dependent on the cellular location (and signaling proteins) of the targeted D2 receptor, a phenomenon termed ‘functional selectivity’ (Kilts et al, 2002; Lawler et al, 1999; Mottola et al, 2002) or ‘agonist trafficking’ (Kenakin, 1995). Whatever the mechanism, aripiprazole has been shown to be effective in treating the positive and negative symptoms of schizophrenia with a low incidence of side effects including minimal short-term weight gain, low liability for inducing movement disorders, and reductions (rather than elevations) in plasma prolactin levels (Goodnick and Jerry, 2002; Goodnick et al, 2002; Kane et al, 2002).
To discover the mechanisms responsible for the favorable actions of aripiprazole in schizophrenia, we conducted a comprehensive pharmacological profiling of aripiprazole at a large number of cloned human molecular targets including GPCRs, ion channels, protein kinases and transporters. We discovered that aripiprazole has a more robust pharmacological profile than previously suspected, including moderate-to-high affinity for a large number of human cloned biogenic amine GPCRs. The current data also demonstrate that aripiprazole can have partial agonist properties at D3-dopamine, and 5-HT1A, 5-HT2A and 5-HT2C serotonin receptors. Finally, consistent with the notion that aripiprazole is not a simple partial agonist, we show that it can appear to be a typical antagonist in many D2L-dopamine functional assays, suggesting the need for further studies on the ‘functional selectivity’ properties of the drug.
METHODS
Radioligand Binding Assays
A large number of transiently and stably transfected cloned human cDNAs, obtained via the resources of the National Institute of Mental Health Psychoactive Drug Screening Program (NIMH-PDSP), were used for radioligand binding and functional assays as previously detailed (Rothman et al, 2000; Tsai et al, 2000). Conditions for radioligand binding assays, along with KD values for standard compounds, are listed in Table 1. In initial screening assays, aripiprazole was tested at a concentration of 10 μM in quadruplicate at a large number of GPCRs, ion channels, and transporters. For molecular targets at which >50% inhibition was measured, Ki determinations were obtained using at least six concentrations of aripiprazole; Ki values were calculated in quadruplicate using GraphPad Prism (GraphPad Software, San Diego California, USA). [125I]DOI competition assays were performed as previously described (Choudhary et al, 1992) with the following changes: 12 dilutions of aripiprazole spanning a range of 0.01–3000 nM were incubated with [125I]DOI (0.3 nM) in total volumes of 0.25 ml at 25°C for 1 h with 5–20 μg of membrane protein in binding buffer (50 mM Tris buffer, pH 7.4, 0.5 mM EDTA, 10 mM MgCl2). Membranes were harvested with a Brandel cell harvester by three ice-cold washes onto polyethyleneimine-pretreated (0.3%) Whatman GF/C filters. Radioactivity bound to filters was quantified by liquid scintillation counting.
PI Hydrolysis Assays
PI hydrolysis experiments were performed as previously described, with minor changes (Bhatnagar et al, 2001; Gray et al, 2001; Shapiro et al, 2000). Briefly, PO1C, GF62, or C6-glioma cells were grown in T75 flasks to 80% confluency, and plated into 24-well plates with Dulbecco's modified Eagle's medium containing 10% dialyzed fetal calf serum. After 24 h, media were aspirated and replaced with a serum-free, inositol-free basal Eagle's medium containing [3H]inositol (1 μCi/ml). After 24 h, media were aspirated and replaced with 1.0 ml of a modified Krebs buffer (118 mM NaCl, 4.7 mM KCl, 2.5 mM CaCl2, 1.2 mM MgCl2, 1.2 mM NaH2PO4, 24 mM NaHCO3, 11 mM dextrose, 18 mM LiCl) preincubated to 37°C and equilibrated with 5% CO2. Seven concentrations of test compounds in triplicate were applied in a total volume of 1.0 ml per well and incubated 1 h. The solution was then quickly aspirated, replaced with 1.0 ml of a stop solution (formic acid/dH2O 1 : 100). After 20 min, the stop solution containing the accumulated inositol phosphates (PIs) was added to columns containing 1 ml of anion exchange resin (formate form) and washed with 12 ml of water followed by 10 ml of 5 mM sodium borate/50 mM sodium formate (Roth et al, 1986). Total PIs were eluted with 10 ml of 0.1 M formic acid/0.2 M ammonium formate into vials containing 3a70B liquid scintillation cocktail (Research Products International, Elk Grove Village, IL), and radioactivity was measure by liquid scintillation counting.
Effects of Aripiprazole on cAMP Production
Inhibition of forskolin-stimulated cAMP production
Inhibition of forskolin-stimulated 3′,5′-cyclic adenosine monophosphate (cAMP) production in stable D4 and 5-HT1A receptor expressing cell lines was measured as previously reported (Lawler et al, 1999; Zhang et al, 1994). In brief, cells were grown in 24-well plates and growth media were replaced with fresh F12 medium containing 100 μM IBMX and 100 μM forskolin (all on ice) just prior to experimentation. Serial dilutions (10-fold) of aripiprazole ranging from 0.1 to 10.000 nM were added to the cells, which were then incubated 20 min at 37°C and 5% CO2. The reaction was terminated by aspiration and the addition of 0.5 ml of ice-cold 3% trichloroacetic acid. Plates were chilled for 1 h at 4°C and spun at 1000 g for 15 min. cAMP was quantified using a competitive binding assay adapted with minor modifications (Nordstedt and Fredholm, 1990). For measurement of cAMP content, trichloroacetic acid extracts (40 μl) were added to reaction tubes containing cAMP assay buffer (100 mM Tris-HCl, pH 7.4, 100 mM NaCl, 5 mM EDTA). [3H]cAMP (1 nM final concentration) was added to each tube, followed by cAMP-binding proteins (approximately 100 μg of crude extract from bovine adrenal cortex in 500 μl of cAMP buffer). The reaction tubes were incubated on ice for 2 h, then harvested with a Brandel cell harvester onto Whatman GF/C filters soaked in water. Filters were allowed to dry, and bound radioactivity was quantified by liquid scintillation counting. The concentration of cAMP in each sample was estimated from a standard curve ranging from 0.1 to 100 pmol of cAMP/assay.
Stimulation of cAMP production
Studies of the effects of serotonin and aripiprazole at 5-HT6 and 5-HT7 receptors were carried out in stable transfectants using methods previously described (Max et al, 1995; Monsma et al, 1993; Shen et al, 1993).
Electrophysiological Studies of D2L, D3, and D4 Receptors
Electrophysiology experiments were performed on MES-23.5 cells stably transfected with D2L, D3, and D4 receptor DNA, and drugs were administered in the bath as previously described (Liu et al, 1999). D2L, D3, and D4 receptor expression levels were 325, 360, and 470 fmol/mg respectively, as originally reported by Liu et al (1999). In brief, cover slips with attached MES-23.5 cell were transferred from 24-well plates into a recording chamber, where cells were perfused continuously at a flow rate of 1–2 ml/min by a solution containing (in mM): NaCl, 135; KCl, 5.4; CaCl2, 1.8; glucose, 20; HEPES, 5; plus 0.5 μM tetrodotoxin, adjusted to pH 7.3 with Tris base, osmolarity adjusted to 320–330 mOsmol. The dish was placed on the stage of an inverted microscope (Zeiss ICM 405) and visualized using Hoffman differential interference contrast optics at × 400. The recording headstage and electrode assembly were held on an articulated arm of a three-position joystick-controlled micromanipulator (MM-8000F, Activational System Inc., Warren, MI) during recording. The voltage-clamp in the whole-cell configuration used an EPC-7/list patch clamp amplifier (Adams-List Associates, Ltd., Westbury, NY). Patch electrodes were fabricated from 1.5 to 1.6 OD MicroHematoctrit capillary tubes (VWR Scientific) pulled in a two-stage process on a Narishigie vertical puller (model PA-81) and polished on a Narishigie microforge (model MF-83). The holding potential and all voltage steps were controlled by PCLAMP software and hardware (Axon Instruments, Foster City, CA) using the CLAMPEX program. The patch electrode solution contained the following (in mM): KCl, 140; MgCl2, 2; CaCl2, 1; HEPES, 10; ATP, 2; cAMP, 0.25; and BAPTA, 0.5; adjusted to pH 7.3 with KOH, osmolarity adjusted to 320–330 mOsmol. When tetraethylammonium (TEA)-Cl (30 mM) was added to the bathing medium, an equimolar concentration of KCl was removed. All solutions were filtered through a 0.45 μM membrane filter. The patch electrode resistance ranged between 2 and 4 MΩ. The seal resistances were greater than 5 GΩ and after membrane rupture the series resistance was between 4.3 and 10.0 MΩ (mean±SEM: 7.1±1.8 megohms; n=100) and the level of capacitance compensation ranged between 10 and 22 pF (mean±SEM and 15.9±6.5 pF; n=100). All recordings were carried out at room temperature (18–22°C). To activate steady-state outward currents, the cells were clamped at a holding potential of −60 mV and stepped to membrane potentials between −80 mV and +90 mV. All drugs were dissolved in the bath medium applied by a perfusion system.
GTPγS Binding Studies
The effects of drugs on [35S]-GTPγ-S binding were determined in the presence and absence of agonist. Nonspecific binding was defined as binding in the presence of 10 μM cold GTPγ-S. Assay tubes contained 150–200 pM [35S]-GTPγ-S, binding buffer (50 mM HEPES, 100 mM NaCl, 4 mM MgCl2, 1 mM EDTA, 0.1% BSA, 0.1% ascorbic acid, pH 7.4 with NaOH), 10 μM GDP and varying concentrations of compounds. CHO hD2L cell membranes (from cells expressing 4.6 pmol/mg receptor) were incubated with test compounds for 15 min at 30°C before addition of [35S]-GTPγ-S. Assay tubes continued to incubate for 30 min. The assay was terminated by filtration with ice-cold wash buffer (50 mM HEPES, 4 mM MgCl2, pH 7.4 with KCl). Filters were allowed to dry, and 20 μl of Packard MicroScint 20 scintillation cocktail was added to each well. Radioactivity in each sample was determined on a Packard Top Count NXT.
Data Analyses
Dose–response curves were analyzed by nonlinear regression using Prism 3.0 software (GraphPad, Inc., San Diego, CA). Other data analysis used InStat 2.0 × (GraphPad, Inc., San Diego, CA) or Prism 3.0 ×. Statistical significance (α=0.05) was determined either by ANOVA followed by the listed post hoc test, or by two-tailed Student's t-test. Some receptor data were analyzed using LIGAND (Munson and Rodbard, 1980).
RESULTS
Affinities of Aripiprazole for Dopamine, Serotonin, and Other Receptors
The affinity of aripiprazole for a variety of receptors was measured by competition radioligand binding assays. Table 1 summarizes the affinity values, and also lists the radioligands and reference compounds used for each assay. The affinities for human 5-HT2A and 5-HT2C receptors were determined for both the inhibition of antagonist ([3H]ketanserin) and agonist ([125I]DOI) radioligands.
Of interest are the relatively high affinities (Ki's 10 nM or less) of aripiprazole for the hD2, and rD3-dopamine, and h5-HT1A−, h5-HT2B−, and h5-HT7 serotonin receptors (Table 1). In addition, aripiprazole had moderate affinity for the hH1 (25.1 nM) and gpH3-histamine (224 nM) receptors, but low affinity (Ki>10 000 nM) for the hH2- and hH4-histamine receptors (Table 1). At all of the adrenergic receptors tested, aripiprazole displayed moderate affinity, highest at the α1A (Ki=25.7 nM) and lowest at α2B (Ki=103 nM). Low affinities were observed at the m1–m5 muscarinic receptors (Ki>1 500 nM).
Of particular interest was the observation that aripiprazole's affinity at D2 receptors was profoundly affected by assay conditions. Thus, when rat D2-receptors were expressed in HEK293 cells using an assay buffer containing NaCl (50 mM Tris-HCl, pH=7.4, 50 mM NaCl, 5 mM MgCl2, 0.5 mM EDTA) affinities were 6.8-fold lower than that reported in rat striatal membranes, but a striking 62-fold lower than those values obtained using stably transfected CHO cells assayed in a Na-free buffer (Lawler et al, 1999). The large effect of sodium ions on the measured affinity of aripiprazole is likely due to the fact that sodium ions decrease agonist affinities for D2-family receptors (Neve, 1991; Neve et al, 1991,2001).
Functional Effects at Serotonin Receptors
The functional characteristics of aripiprazole were assessed in several different cell systems, including some that were also used for receptor screening, as well as others chosen because of their functional utility. The first system studied was the 5-HT1A receptor stably expressed in Chinese Hamster Ovary cells. As shown in Figure 1, serotonin had an EC50 of 4.5 nM, whereas aripiprazole was a low potency (EC50=329 nM) partial agonist at inhibiting forskolin-stimulated cAMP production. At the 5-HT2A receptor (Figure 2), aripiprazole was of even lower intrinsic activity. Thus, in C6-glioma cells that express 5-HT2A receptors without receptor reserve, aripiprazole caused increases in PI hydrolysis only at the highest concentration tested (10 μM). In GF62 cells, a cell line expressing the rat 5-HT2A receptor with substantial receptor reserve, aripiprazole was a partial agonist with an EC50 of 48 nM and intrinsic activity 12.7% that of the full agonist 5-HT.
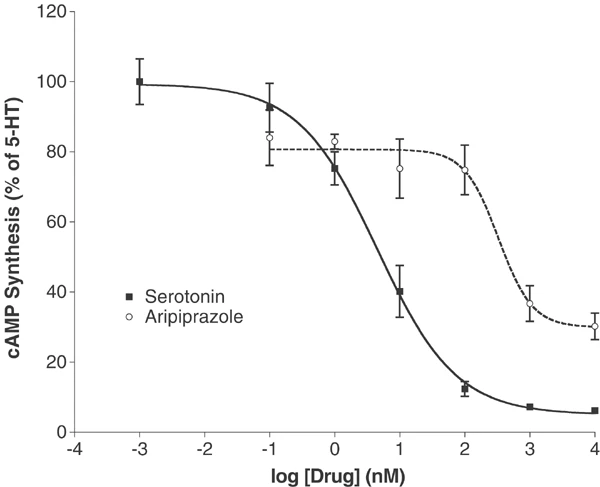
Aripiprazole is a low potency partial agonist at the 5-HT1A receptor stably expressed in 1ACHO cells. Inhibition of forskolin-stimulated cAMP production was compared for serotonin and aripiprazole. Data represent mean±SEM from three separate experiments.
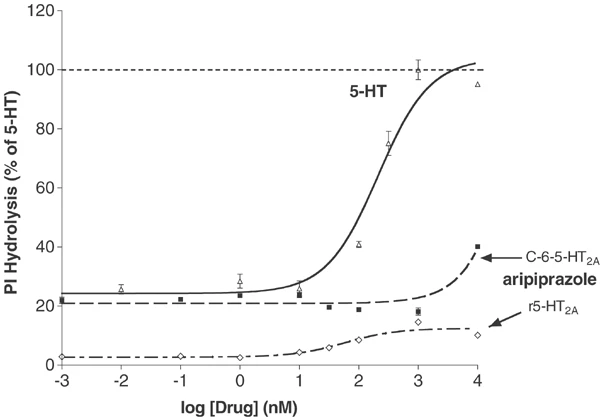
Effect of aripiprazole on serotonin 5-HT2A receptors. Functional effects of aripiprazole on PI hydrolysis were studied in the C-6 glioma line expressing an endogenous rat 5-HT2A receptor (C-6-5-HT2A) and in GF62 cells transfected with the rat 5-HT2A (r5-HT2A). Response data are plotted relative to maximal percentage stimulation by 5-HT in each cell line, with the 5-HT curve being shown in the C-6 line for the sake of clarity. Serotonin-stimulated PI hydrolysis in C-6-glioma cells with an EC50=211 nM, whereas aripiprazole caused partial stimulation only at the highest tested concentration (10 μM). Similarly, in the GF62 cells, aripiprazole was a partial agonist of extremely low intrinsic activity. Data represent mean±SEM of triplicate determinations from three (C-6) or two (GF62) separate experiments.
Next, aripiprazole was studied at 5-HT2B and 5-HT2C receptors. In HEK-293 cells transiently expressing 5-HT2B receptors 5-HT-stimulated PI hydrolysis with an EC50 of 41 nM (Figure 3). At 5-HT2B receptors, aripiprazole was an inverse agonist that decreased PI hydrolysis to approximately half of basal levels, with an EC50 of 11 nM. In contrast, aripiprazole was a partial agonist, albeit with relatively high intrinsic activity, at serotonin 5-HT2C receptors expressed in PO1C cells (r5-HT2c-VGV). As shown in Figure 4, aripiprazole had an EC50 of 26 nM, and an intrinsic activity 82% that of serotonin. Using COS-7 cells transiently transfected with 5-HT2C INI receptor, aripiprazole was a weak full agonist, with an EC50 of 4625±2991 nM and efficacy matching that of the serotonin control (data not shown). The effects of aripiprazole were completely blocked by 100 μM ketanserin (a 5-HT2A/2C antagonist), showing that the effects were indeed 5-HT2C mediated.
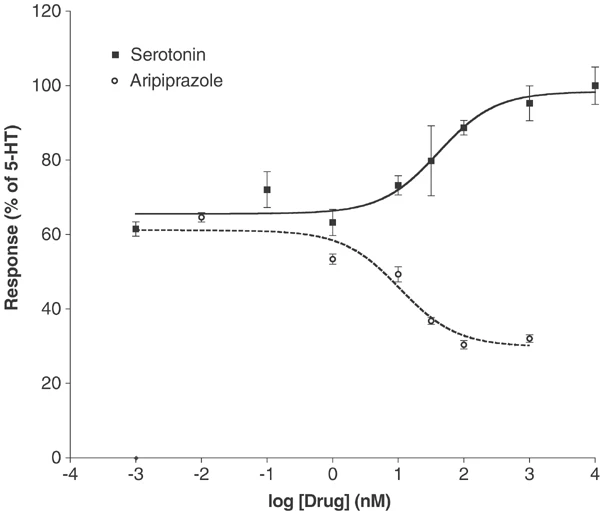
Aripiprazole is an inverse agonist at the 5-HT2B receptor in HEK-293 cells. Serotonin-stimulated PI hydrolysis in HEK 293 cells transiently transfected with 5-HT2B receptors with an EC50 of 41 nM. Aripiprazole decreased PI hydrolysis from a basal level of 61% down to a low of 30% at 1000 nM, with an EC50 of 11 nM. Data represent means±SEM from three separate experiments.

Effect of aripiprazole on serotonin 5-HT2C receptors in PO1C cells. Data represent means and standard errors of triplicate determinations. The single point demonstrates that 100 μM ketanserin can completely block the effects of 10 μM aripiprazole.
The diverse effects caused by aripiprazole were seen again in studies of serotonin 5-HT6 and 5-HT7 receptors. At the 5-HT6 receptor (Figure5, left), serotonin-stimulated cAMP production in 5-HT6 stable transfectants. Aripiprazole (10 μM) had not only no intrinsic activity of its own, but also it blocked essentially all of the effects of serotonin. At the 5-HT7 receptor (Figure 5, right), serotonin, as expected, stimulated cAMP production in 5-HT7 stable transfectants. Here, aripiprazole (10 μM) had barely measurable intrinsic activity, and consistent with this conclusion, the same concentration was able to reduce the effects of serotonin to a level equal to aripiprazole alone. Thus, aripiprazole was an antagonist at 5-HT6 receptors and a weak partial agonist at 5-HT7 receptors.

Antagonist effects of aripiprazole at 5-HT6 and 5-HT7 receptors in stable transfectants. (Left) 5-HT-stimulated cAMP production in 5-HT6 stable transfectants. Aripiprazole (10 μM) blocked essentially all of the effects of serotonin at the 5-HT6 receptor, and had no intrinsic activity of its own. (Right) 5-HT-stimulated cAMP production via 5-HT7 mechanisms, whereas aripiprazole (10 μM) had low intrinsic activity at the 5-HT7 receptor, and effectively blocked the effects of serotonin.
Functional Effects at Dopamine Receptors
Some prior studies have suggested that aripiprazole is a partial agonist at D2 receptors (Burris et al, 2002), while others have proposed that the drug is ‘functionally selective’ at D2 receptors (Lawler et al, 1999). The present experiments thus tested aripiprazole in several different systems, including many not previously studied. The first system studied (Figure 6a) was the MES-23.5 cell line that had stably transfected D2L receptors. As expected, quinpirole caused a 75% increase in outward K+-currents, whereas 10 μM aripiprazole had no effect whatsoever, despite its known affinity for this receptor. A similar pattern was seen when GTPγS binding studies were carried out in D2L-transfected CHO cells (Figure 6b). GTPγS binding was increased markedly by both dopamine and quinpirole, and the D2 antagonist domperidone could block completely the effects of either dopamine or quinpirole. Conversely, aripiprazole alone caused no significant effect, and was equally effective as domperidone at blocking the actions of either dopamine or quinpirole. Finally, in two different cell lines stably expressing the D2L receptor, it was not possible to inhibit forskolin-stimulated cAMP production at any concentration of aripiprazole tested (data not shown). Thus, aripiprazole was a pure antagonist in three different systems expressing the D2L receptor.

Functional effects of aripiprazole at D2L receptors. (a) Quinpirole caused a 75% increase in outward K+-currents in MES-23.5 cells stably transfected with D2L receptors. Conversely, 10 μM aripiprazole had no effect. (b) In D2L-transfected CHO cells, GTPγS binding was increased by both dopamine and quinpirole. Conversely, aripiprazole caused no significant effect on its own, and like the D2 antagonist domperidone, could block the effects of either dopamine or quinpirole. Data represents one of two experiments with identical results.
On the other hand, we replicated the data reported by Burris et al (2002) who found that aripiprazole was a relatively efficacious partial agonist in HEK-293 cells expressing the D2L receptor. As shown in Figure 7, aripiprazole displayed an EC50 of 0.15 nM (pEC50=9.82±0.12) and an intrinsic activity of ca 75% compared to 0.10 nM (10.0±0.05) and 90% for the reference compound bromocriptine. In CHO cells expressing the D2L receptor, aripiprazole had clear agonist activity, although it was less potent (EC50=18.3 nM; pEC50=7.7±1.2) than bromocriptine (EC50=0.12 nM; pEC50=9.9±0.15).

Aripiprazole is a partial agonist in cells stably expressing high levels of the dopamine D2L receptor. HEK 293 and CHO cells stably expressing the dopamine D2 receptor (Burris et al, 2002) were used to assess the agonist actions of aripiprazole. Activities of aripiprazole and the partial agonist bromocriptine were measured using inhibition of forskolin (fsk)-stimulated cAMP production. Values along ordinate represent the cAMP measured at each inhibitor concentration relative to uninhibited, forskolin-stimulated assay wells, normalized to 100%. Data represent mean±SEM of triplicate determinations from one of three replicate experiments having similar results.
MES-23.5 cells were used for studies of both the D3 and D4 receptors. As shown in Figure 8, and in contrast to the effects at D2L receptors in this cell line, both quinpirole (10 μM) and aripiprazole (10 μM) activated D3 receptors. Moreover, the effects of aripiprazole were completely blocked by the D2-like antagonist sulpiride (20 μM). When this same MES-23.5 cell line is transfected with D4 receptors (Figure 9, left), K+ currents were inhibited, rather than stimulated by quinpirole. Aripiprazole (10 μM) caused the same magnitude of effect (ca 30% decrease) as did quinpirole (10 μM). In the same cell line (Figure 9, right), aripiprazole was a partial agonist (EC50=16 nM) at the D4 receptor vs inhibition of forskolin-stimulated cAMP production. Dopamine also caused a dose-dependent inhibition with an EC50=363 nM.

Strong agonist activity of aripiprazole at D3-linked K+-currents in MES-23.5 cells. Quinpirole (10 μM) or aripiprazole (10 μM) both activated D3 receptors, and the effects of aripiprazole were completely blocked by sulpiride.

Differential functional effects of aripiprazole at D4-linked functions. (Left) Aripiprazole (10 μM) and quinpirole (10 μM) caused similar (30–35%) decreases in K+ currents in MES-23.5 cells stably transfected with D4 receptors. (Right) Dopamine and aripiprazole both increased cAMP production in this cell line, although aripiprazole (EC50=16 nM) was of lower intrinsic activity than dopamine (EC50=363 nM). All data represent mean±SEM from three separate experiments.
DISCUSSION
Receptor Profile and Mechanism of Action
While it was known from prior studies that aripiprazole interacted with dopamine and some serotonin receptors (Lawler et al, 1999), the present data show that aripiprazole has a novel and unexpectedly robust pharmacology, displaying functionally significant interactions at a large number of biogenic amine GPCRs including D2-, D3-, and D4-dopamine receptors, and 5-HT1A, 5-HT2A, 5-HT2B, 5-HT2C, 5-HT6 and 5-HT7 serotonin receptors. The current work clearly demonstrates that aripiprazole displays a receptor binding profile substantially distinct from all other approved atypical antipsychotics (ie clozapine, olanzapine, quetiapine, risperidone, and ziprasidone).
Clozapine is now regarded as the prototypical atypical antipsychotic drug, and is the treatment of choice for treatment-resistant and treatment-intolerant schizophrenia and related disorders because of its efficacy and relative lack of EPS. Clozapine treatment also substantially decreases suicidality among patients with schizophrenia, and it may improve negative symptoms, cognition, and tardive dyskinesia (Kane et al, 1988; Meltzer et al, 1989; Meltzer and McGurk, 1999). The molecular mechanism(s) responsible for the actions of clozapine and other related atypical antipsychotics (eg olanzapine, risperidone, quetiapine and ziprasidone) are not proven, but the common view is that it depends on a balanced occupancy of 5-HT2A serotonin and D2-dopamine receptors (Meltzer et al, 1989).
All atypical antipsychotic drugs have a relatively complex in vitro pharmacology with high affinities for a number of serotonin receptors (eg 5-HT1A, 5-HT2A, 5-HT2C, 5-HT6 and 5-HT7 (Milan et al, 1992; Roth et al, 1992,1994). At 5-HT2A and 5-HT2C receptors, most atypical antipsychotic drugs are inverse agonists (Rauser et al, 2001). Atypical antipsychotic drugs also interact, to a greater or lesser extent, with various dopamine receptors including D3-dopamine (Sokoloff et al, 1990) and D4-dopamine (Roth et al, 1995; Van Tol et al, 1991) receptors. At present, it is unclear to what extent binding to receptors other than D2-dopamine and 5-HT2A serotonin receptors contributes to the actions of the atypical antipsychotic drugs. In this regard, it is important to note that aripiprazole has much higher affinity for the human dopamine D2 receptor (0.74 nM) than other atypical antipsychotic drugs, although its affinity is markedly attenuated by sodium (Ki=32.1 nM in the presence of 150 mM NaCl). Of the atypical antipsychotics, only risperidone is reported (in some systems) to show an affinity for the human D2 receptor (0.3–4 nM) approximating that of aripiprazole, while the other atypical antipsychotic drugs have affinities for both rat and human D2 receptors in the range of 3–800 nM (see on-line database at: http://kidb.bioc.cwru.edu/pdsp.php and Arnt and Skarsfeldt, 1998). This is of particular clinical relevance, since blockade of mesolimbic D2 receptors is believed to be related to efficacy against the positive symptoms of schizophrenia (Worrel et al, 2000).
Aripiprazole displayed variable binding affinities for the tested 5-HT receptors, ranging from 0.36 nM (5-HT2B) to 8 μM (5-HT1E) (Table 1). With a Ki=22.4 nM for human 5-HT2A receptors labeled with [3H]ketanserin, aripiprazole had 10–100-fold lower affinity for these receptors than all other tested atypical antipsychotic drugs except quetiapine (31 nM; see on-line database at: http://kidb.bioc.cwru.edu/pdsp.php). Aripiprazole also had low affinity at 5-HT2C receptors labeled with the antagonist [3H]mesulergine (Ki=428 nM), yet much higher affinity for agonist labeled 5-HT2A and 5-HT2C receptors (Table 1). Higher affinity for agonist vs antagonist-labeled 5-HT2- receptors is characteristic of agonists and partial agonists (Egan et al, 2000). This may explain the higher potency aripiprazole has for activating 5-HT2C receptors (23 nM) relative to its inhibition of [3H]mesulergine binding (76 nM).
One of the greatest concerns associated with the use of atypical antipsychotics is the propensity of the drugs to induce weight gain in up to 40% of treated patients (Richelson, 1999; Worrel et al, 2000). Long-term health problems associated with the weight gain (including cardiovascular disease, diabetes, and increased risk of cancer) may be so severe as to contraindicate treatment in some patients. Furthermore, the weight gain may be an important factor in noncompliance. Retrospective studies have concluded that clozapine and olanzapine treatments offer the greatest risk of weight gain, although only clozapine-induced weight gain appeared to be long term and not easily controllable through intervention such as diet and exercise. Of the atypical antipsychotic drugs currently approved for use, risperidone presents an intermediate risk, followed by sertindole and ziprasidone (Wirshing et al, 1999). The greatest single correlate for weight gain has been antagonism of the H1-histamine receptor (Wirshing et al, 1999; Kroeze et al, 2003). The moderate binding affinity of aripiprazole for the human H1-histamine receptor (25.1 nM) predicts that aripiprazole will exhibit a minimal propensity to induce short-term weight gain (Kroeze et al, 2003). The results of ongoing clinical trials, however, should answer this question conclusively.
Functional Effects at Target Receptors and the ‘Functional Selectivity Hypothesis’
D2-receptors
The functional findings in the current work are of particular importance, since the actions of aripiprazole differ markedly across receptor systems. Thus, in the present studies, aripiprazole was sometimes an antagonist (eg at 5-HT6 and D2L), sometimes an inverse agonist (eg 5-HT2B), sometimes a partial agonist (eg D2L), and sometimes a full agonist (D3, D4). Aripiprazole was frequently found to be a partial agonist, with an intrinsic activity that could be low (D2L, 5-HT2A, 5-HT7), intermediate (5-HT1A), or high (D4, 5-HT2C). We recognize (vide infra) that in vitro assay systems may not accurately reflect the actions of the drug in vivo, yet we feel the present data do provide important clues as to the mechanism of action of aripiprazole. This mixture of agonist actions at D2-dopamine receptors is consistent with the hypothesis proposed by Lawler et al (1999) that aripiprazole has ‘functionally selective’ actions. The ‘functional-selectivity’ hypothesis proposes that, depending upon the cellular milieu (eg receptor and G protein complement and concentration), a mixture of agonist/partial agonist/antagonist actions are likely (Kilts et al, 2002; Lawler et al, 1999; Mottola et al, 2002). According to this hypothesis, agonists may induce structural changes in receptor conformations that are differentially ‘sensed’ by the local complement of G proteins to induce a variety of functional actions depending upon the precise cellular milieu. The diverse actions of aripiprazole at D2-dopamine receptors are clearly cell-type specific (eg agonism, antagonism, partial agonism), and are most parsimoniously explained by the ‘functional selectivity’ hypothesis.
Indeed, the functional effects of aripiprazole have been extensively studied at dopamine D2 receptors. Prior studies have supported the notion that the unique clinical actions of aripiprazole are the result of its partial agonist properties at the D2L receptor (Burris et al, 2002; Inoue et al, 1996; Tamminga, 2002). Burris et al (2002) have shown that in CHO cells transfected with hD2L (500–1000 fmol/mg; see Filtz et al, 1993), aripiprazole behaves as a classical partial agonist. In this study, we were able to replicate the Burris et al (2002) finding that aripiprazole has partial agonist actions when using the identical cell lines. Conversely, others (Lawler et al, 1999) concluded that the functional properties of aripiprazole are affected markedly by cell type and other variables to a much greater degree than by other drugs. Thus, Lawler et al (1999) hypothesized that a mechanism called ‘functional selectivity’/‘agonist trafficking’ might be critical to the actions at the D2L receptor in brain. Specifically, they suggested that the functional characteristics of aripiprazole at a single receptor isoform (eg the D2L) are highly dependent on the location of the receptor (ie influenced by the signaling partners such as G proteins). The current studies report data from a variety of systems in which aripiprazole has not only no detectable intrinsic activity, but in one of the systems is a complete antagonist of both dopamine and quinpirole. It is unlikely that the differential actions of aripiprazole as an agonist, antagonist, or partial agonist were entirely due to differences in relative D2 receptor expression since aripiprazole was an antagonist in cells with the highest level of expression 4.6 pmol/mg) and a partial agonist in cells with an intermediate level of expression (0.5–1 pmol/mg). Instead, the current data are most parsimoniously explained by the ‘functional selectivity’ hypothesis of Lawler et al (1999).
5-HT2A/B/C receptors
Since 5-HT2C receptors have been implicated in the control of depression, OCD, and appetite, agonism at the 5-HT2C receptor might be associated with therapeutic potential in obsessive compulsive disorder, obesity, and depression (Martin et al, 1998). 5-HT2C agonism has been demonstrated to induce anorexia via enhancement of serotonergic neurotransmission via activation of 5-HT2C receptors (Vickers et al, 1999); it is conceivable that the 5-HT2C agonist actions of aripiprazole may, thus, be partly responsible for the minimal weight gain associated with this compound in clinical trials. In terms of potential action as an antiobsessional agent, it is worthwhile noting that a variety of 5-HT2A/5-HT2C agonists have shown promise as antiobsessional agents, yet many of these compounds are hallucinogenic, presumably due to 5-HT2A activation (Roth et al, 1999). Based on data obtained using C6-glioma cells, which express 5-HT2A receptors with minimal receptor reserve, we conclude that aripiprazole will function as a 5-HT2A antagonist in vivo. Thus, aripiprazole has a favorable pharmacological profile in being a 5-HT2A antagonist and a 5-HT2C partial agonist. Based on this profile, one can predict that aripiprazole may have antiobsessional and anorectic actions in humans.
The other member of the 5-HT2-receptor family that aripiprazole has high affinity for is the 5-HT2B receptor. Since agonism at 5-HT2B receptors is associated with vavlular heart disease (Fitzgerald et al, 2000; Rothman et al, 2000), it is important to determine whether aripiprazole was an agonist or antagonist at 5-HT2B receptors. The current data demonstrate that aripiprazole is an inverse agonist at 5-HT2B receptors, and is thus unlikely to induce valvular heart disease. On the other hand, 5-HT2B antagonism has been promoted as a pharmacologic approach for treating migraine headadches (Hamel, 1999; Johnson et al, 1998). This is because of the observation that, during a migraine attack, activation of 5-HT2B receptors has been reported to accompany a sudden rise in serotonin, potentially resulting in activation of sensory neurons and the onset of pain. It will be interesting to see whether the potent antagonism (inverse agonism) of aripiprazole at the 5-HT2B receptor will translate into an effective antimigraine therapy.
The prefrontal cortex contains large densities of both 5-HT1A and 5-HT2A receptors, located primarily on pyramidal neurons. As cortical 5-HT2A receptors are excitatory, and cortical 5-HT1A receptors are inhibitory, it has been suggested recently that blockade of cortical 5-HT2A receptors and/or functional 5-HT1A receptor agonism may mediate the physiological balance between excitatory and inhibitory inputs onto prefrontal pyramidal neurons (Martin-Ruiz et al, 2001). In preclinical trials, 5-HT1A selective agonists were capable of antagonizing (attenuating) neuroleptic-induced EPS in animal models (Ellenbroek et al, 1994; Liebman et al, 1989). Based on this finding, Feenstra et al (2001) examined the structure/affinity relationship of a large panel of 5-HT1A receptor binding an analogs. At least one of these analogs shows promise as an antipsychotic drug in rodents and is being developed for pharmacological study. Of the current atypical antipsychotic drugs, ziprasidone is the only drug that binds with similar affinity to aripiprazole at the 5-HT1A receptor. Thus, 5-HT1A partial agonism is not a common pharmacological characteristic of atypical antipsychotic drugs. It remains to be seen, then, whether 5-HT1A selective agonism, alone or in combination with effective 5-HT2A antagonism, will be efficacious in treating the various symptoms of schizophrenia.
It is nonetheless clear that aripiprazole is an atypical antipsychotic drug with a unexpectedly robust pharmacology, and, thus, shows promise as the first in a new generation of atypical antipsychotic drugs. The pharmacological profile of aripiprazole is consistent with a favorable in vivo pharmacology in humans including a relatively low propensity to induce either weight gain or EPS. It is also possible that aripiprazole may prove to be effective in other disorders based on the unusually broad pharmacologic profile uncovered in this survey. The current data suggest that the new class of drugs represented by aripiprazole may work by being either dopamine-serotonin partial agonistş and/or by working through the mechanism of ‘functional selectivity’/‘agonist trafficking’ (eg at the D2 and possibly some 5-HT receptors). Our research clearly points to the need for further studies on both multiple receptor targeting and ‘functional selectivity’/‘agonist trafficking.’ Such mechanisms also open the door for other therapeutic indications for aripiprazole, including its use as a treatment for migraine, obesity, obsessive-compulsive disorder and other psychiatric conditions.
References
Arnt J, Skarsfeldt T (1998). Do novel antipsychotics have similar pharmacological characteristics? A review of the evidence. Neuropsychopharmacology 18: 63–101.
Bhatnagar A, Willins DL, Gray JA, Woods J, Benovic JL, Roth BL (2001). The dynamin-dependent, arrestin-independent internalization of 5-hydroxytryptamine 2A (5-HT2A) serotonin receptors reveals differential sorting of arrestins and 5-HT2A receptors during endocytosis. J Biol Chem 276: 8269–8277.
Burris KD, Molski TF, Xu C, Ryan E, Tottori K, Kikuchi T et al (2002). Aripiprazole, a novel antipsychotic, is a high-affinity partial agonist at human dopamine D2 receptors. J Pharmacol Exp Ther 302: 381–389.
Choudhary MS, Craigo S, Roth BL (1992). Identification of receptor domains that modify ligand binding to 5-hydroxytryptamine2 and 5-hydroxytryptamine1c serotonin receptors. Mol Pharmacol 42: 627–633.
Creese I, Burt DR, Snyder SH (1976). Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Science 192: 481–483.
Egan C, Grinde E, Dupre A, Roth BL, Hake M, Teitler M et al (2000). Agonist high and low affinity state ratios predict drug intrinsic activity and a revised ternary complex mechanism at serotonin 5-HT(2A) and 5-HT(2C) receptors. Synapse 35: 144–150.
Ellenbroek BA, Prinssen EP, Cools AR (1994). The role of serotonin receptor subtypes in the behavioural effects of neuroleptic drugs. A paw test study in rats. Eur J Neurosci 6: 1–8.
Feenstra RW, de Moes J, Hofma JJ, Kling H, Kuipers W, Long SK et al (2001). New 1-aryl-4-(biarylmethylene)piperazines as potential atypical antipsychotics sharing dopamine D(2)-receptor and serotonin 5-HT(1A)-receptor affinities. Bioorg Med Chem Lett 11: 2345–2349.
Filtz TM, Artymyshyn RP, Guan W, Molinoff PB (1993). Paradoxical regulation of dopamine receptors in transfected 293 cells. Mol Pharmacol 44: 371–379.
Fitzgerald LW, Burn TC, Brown BS, Patterson JP, Corjay MH, Valentine PA et al (2000). Possible role of valvular serotonin 5-HT(2B) receptors in the cardiopathy associated with fenfluramine. Mol Pharmacol 57: 75–81.
Ghaemi SN, Cherry EL, Katzow JA, Goodwin FK (2000). Does olanzapine have antidepressant properties? A retrospective preliminary study. Bipolar Disord 2: 196–199.
Goodnick PJ, Jerry JM (2002). Aripiprazole: profile on efficacy and safety. Expert Opin Pharmacother 3: 1773–1781.
Goodnick PJ, Rodriguez L, Santana O (2002). Antipsychotics: impact on prolactin levels. Expert Opin Pharmacother 3: 1381–1391.
Gray JA, Sheffler DJ, Bhatnagar A, Woods JA, Hufeisen SJ, Benovic JL et al (2001). Cell-type specific effects of endocytosis inhibitors on 5-hydroxytryptamine(2A) receptor desensitization and resensitization reveal an arrestin-, GRK2-, and GRK5-independent mode of regulation in human embryonic kidney 293 cells. Mol Pharmacol 60: 1020–1030.
Hamel E (1999). The biology of serotonin receptors: focus on migraine pathophysiology and treatment. Can J Neurol Sci 26(Suppl 3): S2–S6.
Inoue A, Miki S, Seto M, Kikuchi T, Morita S, Ueda H et al (1997). Aripiprazole, a novel antipsychotic drug, inhibits quinpirole-evoked GTPase activity but does not up-regulate dopamine D2 receptor following repeated treatment in the rat striatum. Eur J Pharmacol 321: 105–111.
Inoue T, Domae M, Yamada K, Furukawa T (1996). Effects of the novel antipsychotic agent 7-(4-[4-(2,3-dichlorophenyl)-1-piperazinyl]butyloxy)-3,4-dihydro-2(1H)-quinolinone (OPC-14597) on prolactin release from the rat anterior pituitary gland. J Pharmacol Exp Ther 277: 137–143.
Johnson KW, Phebus LA, Cohen ML (1998). Serotonin in migraine: theories, animal models and emerging therapies. Prog Drug Res 51: 219–244.
Kane J, Honigfeld G, Singer J, Meltzer H (1988). Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry 45: 789–796.
Kane JM, Carson WH, Saha AR, McQuade RD, Ingenito GG, Zimbroff DL et al (2002). Efficacy and safety of aripiprazole and haloperidol versus placebo in patients with schizophrenia and schizoaffective disorder. J Clin Psychiatry 63: 763–771.
Kenakin T (1995). Agonist-receptor efficacy. II. Agonist trafficking of receptor signals. Trends Pharmacol Sci 16: 232–238.
Kilts JD, Connery HS, Arrington EG, Lewis MM, Lawler CP, Oxford GS et al (2002). Functional selectivity of dopamine receptor agonists. II. Actions of dihydrexidine in D2L receptor-transfected MN9D cells and pituitary lactotrophs. J Pharmacol Exp Ther 301: 1179–1189.
Kroeze WK, Hufeisen SJ, Popadak BA, Renock SM, Steinberg S, Ernsberger P et al (2003). H1-histamine receptor affinity predicts short-term weight gain for typical and atypical antipsychotic drugs. Neuropsychopharmacology 28(3): 519–526.
Lawler CP, Prioleau C, Lewis MM, Mak C, Jiang D, Schetz JA et al (1999). Interactions of the novel antipsychotic aripiprazole (OPC-14597) with dopamine and serotonin receptor subtypes. Neuropsychopharmacology 20: 612–627.
Liebman JM, Gerhardt SC, Gerber R (1989). Effects of 5-HT1A agonists and 5-HT2 antagonists on haloperidol-induced dyskinesias in squirrel monkeys: no evidence for reciprocal 5-HT- dopamine interaction. Psychopharmacology 97: 456–461.
Liu LX, Burgess LH, Gonzalez AM, Sibley DR, Chiodo LA (1999). D2S, D2L, D3, and D4 dopamine receptors couple to a voltage-dependent potassium current in N18TG2 × mesencephalon hybrid cell (MES-23.5) via distinct G proteins. 31: 108–118.
Martin JR, Bos M, Jenck F, Moreau J, Mutel V, Sleight AJ et al (1998). 5-HT2C receptor agonists: pharmacological characteristics and therapeutic potential. J Pharmacol Exp Ther 286: 913–924.
Martin-Ruiz R, Puig MV, Celada P, Shapiro DA, Roth BL, Mengod G et al (2001). Control of serotonergic function in medial prefrontal cortex by serotonin-2A receptors through a glutamate-dependent mechanism. J Neurosci 21: 9856–9866.
Max SI, Monsma Jr FJ, Sibley DR (1995). Agonist-induced desensitization of 5-HT6 serotonin receptor-coupled adenylyl cyclase in stably transfected HEK-293 cells. J Serotonin Res 2: 101–116.
Meltzer HY, Matsubara S, Lee JC (1989). Classification of typical and atypical antipsychotic drugs on the basis of dopamine D-1, D-2 and serotonin2 pKi values. J Pharmacol Exp Ther 251: 238–246.
Meltzer HY, McGurk SR (1999). The effects of clozapine, risperidone, and olanzapine on cognitive function in schizophrenia. Schizophr Bull 25: 233–255.
Milan N, Brussels J, Milan G (1992). New generation of antipsychotic drugs: novel mechanisms of action. Workshop, Monte Carlo, March 16–18, 1992. pp 1–14.
Monsma Jr FJ, Shen Y, Ward RP, Hamblin MW, Sibley DR (1993). Cloning and expression of a novel serotonin receptor with high affinity for tricyclic psychotropic drugs. Mol Pharmacol 43: 320–327.
Mottola DM, Kilts JD, Lewis MM, Connery HS, Walker QD, Jones SR et al (2002). Functional selectivity of dopamine receptor agonists. I. Selective activation of postsynaptic dopamine D2 receptors linked to adenylate cyclase. J Pharmacol Exp Ther 301: 1166–1178.
Munson PJ, Rodbard D (1980). LIGAND: a versatile, computerized approach for characterization of ligand-binding systems. Anal Biochem 107: 220–239.
Narendran R, Young CM, Valenti AM, Pristach CA, Pato MT, Grace JJ (2001). Olanzapine therapy in treatment-resistant psychotic mood disorders: a long-term follow-up study. J Clin Psychiatry 62: 509–516.
Neve KA (1991). Regulation of dopamine D2 receptors by sodium and pH. Mol Pharmacol 39: 570–578.
Neve KA, Cox BA, Henningsen RA, Spanoyannis A, Neve RL (1991). Pivotal role for aspartate-80 in the regulation of dopamine D2 receptor affinity for drugs and inhibition of adenylyl cyclase. Mol Pharmacol 39: 733–739.
Neve KA, Cumbay MG, Thompson KR, Yang R, Buck DC, Watts VJ et al (2001). Modeling and mutational analysis of a putative sodium-binding pocket on the dopamine D2 receptor. Mol Pharmacol 60: 373–381.
Nordstedt C, Fredholm BB (1990). A modification of a protein-binding method for rapid quantification of cAMP in cell-culture supernatants and body fluid. Anal Biochem 189: 231–234.
Oshiro Y, Sato S, Kurahashi N, Tanaka T, Kikuchi T, Tottori K et al (1998). Novel antipsychotic agents with dopamine autoreceptor agonist properties: synthesis and pharmacology of 7-[4-(4-phenyl-1-piperazinyl)butoxy]-3,4-dihydro-2(1H)-quinolinone derivatives. J Med Chem 41: 658–667.
Rauser L, Savage JE, Meltzer HY, Roth BL (2001). Inverse agonist actions of typical and atypical antipsychotic drugs at the human 5-hydroxytryptamine(2C) receptor. J Pharmacol Exp Ther 299: 83–89.
Richelson E (1999). Receptor pharmacology of neuroleptics: relation to clinical effects. J Clin Psychiatry 60: 5–14.
Roth BL (1994). Multiple serotonin receptors: clinical and experimental aspects. Ann Clin Psychiatry 6: 67–78.
Roth BL (2000). Neuronal signal transduction pathways: wasteland or the promised land? Sci STKE 2000(45): PE1.
Roth BL, Ciaranello RD, Meltzer HY (1992). Binding of typical and atypical antipsychotic agents to transiently expressed 5-HT1C receptors. J Pharmacol Exp Ther 260: 1361–1365.
Roth BL, Craigo SC, Choudhary MS, Uluer A, Monsma Jr FJ, Shen Y et al (1994). Binding of typical and atypical antipsychotic agents to 5-hydroxytryptamine-6 and 5-hydroxytryptamine-7 receptors. J Pharmacol Exp Ther 268: 1403–1410.
Roth BL, Meltzer H, Khan N (1998). Binding of typical and atypical antipsychotic drugs to multiple neurotransmitter receptors. In: August JT, Anders MW, Murad F, Coyle JT (eds). Advances in Pharmacology, Vol 42 Academic Press: San Diego. pp 482–485.
Roth BL, Nakaki T, Chuang DM, Costa E (1986). 5-Hydroxytryptamine2 receptors coupled to phospholipase C in rat aorta: modulation of phosphoinositide turnover by phorbol ester. J Pharmacol Exp Ther 238: 480–485.
Roth BL, Sheffler D, Potkin S (In Press). Atypical antipsychotic drug actions: unitary or multiple mechanisms for ‘Atypicality’? Clin Neurosci Res.
Roth BL, Tandra S, Burgess LH, Sibley DR, Meltzer HY (1995). D4 dopamine receptor binding affinity does not distinguish between typical and atypical antipsychotic drugs. Psychopharmacology (Berl) 120: 365–368.
Roth BL, Willins D, Kristiansen K, Kroeze W (1999). Activation is hallucinogenic and antagonism is therapeutic: role of 5-HT2A receptors in atypical antipsychotic drug actions. Neuroscientist 5: 254–262.
Rothman RB, Baumann MH, Savage JE, Rauser L, McBride A, Hufeisen SJ et al (2000). Evidence for possible involvement of 5-HT(2B) receptors in the cardiac valvulopathy associated with fenfluramine and other serotonergic medications. Circulation 102: 2836–2841.
Sanger TM, Grundy SL, Gibson PJ, Namjoshi MA, Greaney MG, Tohen MF (2001). Long-term olanzapine therapy in the treatment of bipolar I disorder: an open-label continuation phase study. J Clin Psychiatry 62: 273–281.
Shapiro DA, Kristiansen K, Kroeze WK, Roth BL (2000). Differential modes of agonist binding to 5-hydroxytryptamine(2A) serotonin receptors revealed by mutation and molecular modeling of conserved residues in transmembrane region 5. Mol Pharmacol 58: 877–886.
Shen Y, Monsma Jr FJ, Metcalf MA, Jose PA, Hamblin MW, Sibley DR (1993). Molecular cloning and expression of a 5-hydroxytryptamine7 serotonin receptor subtype. J Biol Chem 268: 18200–18204.
Sokoloff P, Giros B, Martres MP, Bouthenet ML, Schwartz JC (1990). Molecular cloning and characterization of a novel dopamine receptor (D3) as a target for neuroleptics. Nature 347: 146–151.
Stanton T, Bolden-Watson C, Cusack B, Richelson E (1993). Antagonism of the five cloned human muscarinic cholinergic receptors expressed in CHO-K1 cells by antidepressants and antihistaminics. Biochem Pharmacol 45: 2352–2354.
Tamminga CA (2002). Partial dopamine agonists in the treatment of psychosis. J Neural Transm 109: 411–420.
Tsai Y, Dukat M, Slassi A, MacLean N, Demchyshyn L, Savage JE et al (2000). N1-(Benzenesulfonyl)tryptamines as novel 5-HT6 antagonistst. Bioorg Med Chem Lett 10: 2295–2299.
Van Tol HHM, Bunzow JR, Guan HC, Sunahara H-C, Seeman P, Niznik HB et al (1991). Cloning of the gene for a human dopamine D4 receptor with high affinity for the antipsychotic clozapine. Nature 350: 610–614.
Vickers SP, Clifton PG, Dourish CT, Tecott LH (1999). Reduced satiating effect of d-fenfluramine in serotonin 5-HT(2C) receptor mutant mice. Psychopharmacology (Berl) 143: 309–314.
Wirshing DA, Wirshing WC, Kysar L, Berisford MA, Goldstein D, Pashdag J et al (1999). Novel antipsychotics: comparison of weight gain liabilities. J Clin Psychiatry 60: 358–363.
Worrel JA, Marken PA, Beckman SE, Ruehter VL (2000). Atypical antipsychotic agents: a critical review. Am J Health Syst Pharm 57: 238–255.
Wyatt RJ, Henter I, Leary MC, Taylor E (1995). An economic evaluation of schizophrenia—1991. Soc Psychiatry Psychiatr Epidemiol 30: 196–205.
Zeng XP, Le F, Richelson E (1997). Muscarinic m4 receptor activation by some atypical antipsychotic drugs. Eur J Pharmacol 321: 349–354.
Zhang LJ, Lachowicz JE, Sibley DR (1994). The D2S and D2L dopamine receptor isoforms are differentially regulated in Chinese hamster ovary cells. Mol Pharmacol 45: 878–889.
Acknowledgements
DAS was supported in part by a NARSAD Young Investigator Award. Additional support was provided by the NIMH Psychoactive Drug Screening Program NO2MH80005 to BLR, by an NIMH Research Scientist Development Award KO2MH01366 to BLR, and by MH40537 and MH53356 to RBM.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Shapiro, D., Renock, S., Arrington, E. et al. Aripiprazole, A Novel Atypical Antipsychotic Drug with a Unique and Robust Pharmacology. Neuropsychopharmacol 28, 1400–1411 (2003). https://doi.org/10.1038/sj.npp.1300203
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.npp.1300203
Keywords
This article is cited by
-
Pharmacological treatment of Tourette’s syndrome: from the past to the future
Neurological Sciences (2024)
-
SSELM-neg: spherical search-based extreme learning machine for drug–target interaction prediction
BMC Bioinformatics (2023)
-
The Sapap3−/− mouse reconsidered as a comorbid model expressing a spectrum of pathological repetitive behaviours
Translational Psychiatry (2023)
-
Genome-wide association study identified six loci associated with adverse drug reactions to aripiprazole in schizophrenia patients
Schizophrenia (2023)
-
Analysis of neurotransmitters validates the importance of the dopaminergic system in autism spectrum disorder
World Journal of Pediatrics (2023)
