
Abstract
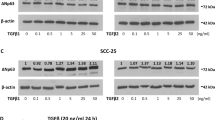
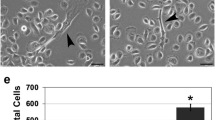
Recent studies indicate that the specificity of p38 mitogen-activated protein kinase (MAPK)-mediated cellular stress responses is determined by the expression pattern of the distinct p38 isoforms. Here, we have analysed the function of distinct p38 isoforms in the growth and invasion of head and neck squamous cell carcinomas (HNSCCs). Activation of p38 MAPK by arsenite resulted in inactivation of the ERK1,2 signaling pathway by dephosphorylation of MEK1,2 in primary human epidermal keratinocytes (HEKs), whereas in HNSCC cells this p38-mediated inhibition of the ERK1,2 pathway was absent. Quantitation of p38 pathway component mRNA expression in HNSCC cell lines (n=42) compared to HEKs (n=8) revealed that p38α and p38δ isoforms are predominantly expressed in both cell types and that MKK3 is the primary upstream activator expressed. Inhibition of endogenous p38α or p38δ activity by adenoviral delivery of corresponding dominant-negative p38 isoforms potently reduced MMP-13 and MMP-1 expressions, and suppressed the invasion of HNSCC cells through collagen. Dominant-negative p38α and p38δ inhibited squamous cell carcinoma (SCC) cell proliferation and inhibition of p38α activity also compromised survival of SCC cells. p38α and p38δ were predominantly expressed in HNSCCs (n=24) and nonneoplastic epithelium in vivo (n=6), with MKK3 being the primary upstream activator. Activation and expression of p38α and p38δ by tumor cells was detected in HNSCCs in vivo (n=16). Adenoviral expression of dominant-negative p38α or p38δ in cutaneous SCC cells potently inhibited their implantation in skin of severe combined immunodeficiency mice and growth of xenografts in vivo. Our results indicate that p38α and p38δ specifically promote the malignant phenotype of SCC cells by regulating cell survival, proliferation and invasion, suggesting these p38 MAPK isoforms as potential therapeutic targets in HNSCCs.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 50 print issues and online access
$259.00 per year
only $5.18 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others

References
Aguirre-Ghiso JA, Liu D, Mignatti A, Kovalski K, Ossowski L . (2001). Urokinase receptor and fibronectin regulate the ERK(MAPK) to p38(MAPK) activity ratios that determine carcinoma cell proliferation or dormancy in vivo. Mol Biol Cell 12: 863–879.
Ala-aho R, Ahonen M, George SJ, Heikkilä J, Grenman R, Kallajoki M et al. (2004). Targeted inhibition of human collagenase-3 (MMP-13) expression inhibits squamous cell carcinoma growth in vivo. Oncogene 23: 5111–5123.
Ala-aho R, Grénman R, Seth P, Kähäri VM . (2002). Adenoviral delivery of p53 gene suppresses expression of collagenase-3 (MMP-13) in squamous carcinoma cells. Oncogene 21: 1187–1195.
Ambrosino C, Mace G, Galban S, Fritsch C, Vintersten K, Black E et al. (2003). Negative feedback regulation of MKK6 mRNA stability by p38α mitogen-activated protein kinase. Mol Cell Biol 23: 370–381.
Boyce ST, Ham RG . (1983). Calcium-regulated differentiation of normal human epidermal keratinocytes in chemically defined clonal culture and serum-free serial culture. J Invest Dermatol 81 (Suppl. 1): 33s–40s.
Brachman DG, Graves D, Vokes E, Beckett M, Haraf D, Montag A et al. (1992). Occurrence of p53 gene deletions and human papilloma virus infection in human head and neck cancer. Cancer Res 52: 4832–4836.
Bulavin DV, Demidov ON, Saito S, Kauraniemi P, Phillips C, Amundson SA et al. (2002). Amplification of PPM1D in human tumors abrogates p53 tumor-suppressor activity. Nat Genet 31: 210–215.
Bulavin DV, Saito S, Hollander MC, Sakaguchi K, Anderson CW, Appella E et al. (1999). Phosphorylation of human p53 by p38 kinase coordinates N-terminal phosphorylation and apoptosis in response to UV radiation. EMBO J 18: 6845–6854.
Chomczynski P, Sacchi N . (1987). Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 162: 156–159.
Efimova T, Broome AM, Eckert RL . (2003). A regulatory role for p38δ MAPK in keratinocyte differentiation. Evidence for p38δ-ERK1/2 complex formation. J Biol Chem 278: 34277–34285.
Egeblad M, Werb Z . (2002). New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer 2: 161–174.
Enslen H, Brancho DM, Davis RJ . (2000). Molecular determinants that mediate selective activation of p38 MAP kinase isoforms. EMBO J 19: 1301–1311.
Enslen H, Raingeaud J, Davis RJ . (1998). Selective activation of p38 mitogen-activated protein (MAP) kinase isoforms by the MAP kinase kinases MKK3 and MKK6. J Biol Chem 273: 1741–1748.
Garrington TP, Johnson GL . (1999). Organization and regulation of mitogen-activated protein kinase signaling pathways. Curr Opin Cell Biol 11: 211–218.
Hale KK, Trollinger D, Rihanek M, Manthey CL . (1999). Differential expression and activation of p38 mitogen-activated protein kinase α, β, γ and δ in inflammatory cell lineages. J Immunol 162: 4246–4252.
Hanahan D, Weinberg RA . (2000). The hallmarks of cancer. Cell 100: 57–70.
Huang S, New L, Pan Z, Han J, Nemerow GR . (2000). Urokinase plasminogen activator/urokinase-specific surface receptor expression and matrix invasion by breast cancer cells requires constitutive p38α mitogen-activated protein kinase activity. J Biol Chem 275: 12266–12272.
Hunter KD, Parkinson EK, Harrison PR . (2005). Profiling early head and neck cancer. Nat Rev Cancer 5: 127–135.
Johansson N, Airola K, Grenman R, Kariniemi AL, Saarialho-Kere U, Kähäri VM . (1997). Expression of collagenase-3 (matrix metalloproteinase-13) in squamous cell carcinomas of the head and neck. Am J Pathol 151: 499–508.
Johansson N, Ala-aho R, Uitto V, Grenman R, Fusenig NE, Lopez-Otin C et al. (2000). Expression of collagenase-3 (MMP-13) and collagenase-1 (MMP-1) by transformed keratinocytes is dependent on the activity of p38 mitogen-activated protein kinase. J Cell Sci 113: 227–235.
Keesler GA, Bray J, Hunt J, Johnson DA, Gleason T, Yao Z et al. (1998). Purification and activation of recombinant p38 isoforms α, β, γ, and δ. Protein Expr Purif 14: 221–228.
Lansdorf CD, Grénman R, Bier H, Somers KD, Kim SY, Whiteside TL et al. (1999). Head and neck cancers. In: Masters J, Palsson B (eds). Human Cell Culture, Vol 2, Cancer Cell Lines Part 2. Kluwer Academic Press: Dordrecht, Holland, pp 185–255.
Li SP, Junttila MR, Han J, Kähäri VM, Westermarck J . (2003). p38 Mitogen-activated protein kinase pathway suppresses cell survival by inducing dephosphorylation of mitogen-activated protein/extracellular signal-regulated kinase kinase1,2. Cancer Res 63: 3473–3477.
Liu Q, Hofmann PA . (2004). Protein phosphatase 2A-mediated cross-talk between p38 MAPK and ERK in apoptosis of cardiac myocytes. Am J Physiol Heart Circ Physiol 286: H2204–H2212.
Olson JM, Hallahan AR . (2004). p38 MAP kinase: a convergence point in cancer therapy. Trends Mol Med 10: 125–129.
Ono K, Han J . (2000). The p38 signal transduction pathway: activation and function. Cell Signal 12: 1–13.
Pramanik R, Qi X, Borowicz S, Choubey D, Schultz RM, Han J et al. (2003). p38 isoforms have opposite effects on AP-1-dependent transcription through regulation of c-Jun. The determinant roles of the isoforms in the p38 MAPK signal specificity. J Biol Chem 278: 4831–4839.
Pruitt K, Pruitt WM, Bilter GK, Westwick JK, Der CJ . (2002). Raf-independent deregulation of p38 and JNK mitogen-activated protein kinases are critical for Ras transformation. J Biol Chem 277: 31808–31817.
Ravanti L, Häkkinen L, Larjava H, Saarialho-Kere U, Foschi M, Han J et al. (1999). Transforming growth factor-β induces collagenase-3 expression by human gingival fibroblasts via p38 mitogen-activated protein kinase. J Biol Chem 274: 37292–37300.
Reddy KB, Nabha SM, Atanaskova N . (2003). Role of MAP kinase in tumor progression and invasion. Cancer Metastasis Rev 22: 395–403.
Reunanen N, Li SP, Ahonen M, Foschi M, Han J, Kähäri VM . (2002). Activation of p38α MAPK enhances collagenase-1 (matrix metalloproteinase (MMP)-1) and stromelysin-1 (MMP-3) expression by mRNA stabilization. J Biol Chem 277: 32360–32368.
Simon C, Goepfert H, Boyd D . (1998). Inhibition of the p38 mitogen-activated protein kinase by SB 203580 blocks PMA-induced Mr 92 000 type IV collagenase secretion and in vitro invasion. Cancer Res 58: 1135–1139.
Somers KD, Merrick MA, Lopez ME, Incognito LS, Schechter GL, Casey G . (1992). Frequent p53 mutations in head and neck cancer. Cancer Res 52: 5997–6000.
Vihinen P, Kähäri VM . (2002). Matrix metalloproteinases in cancer: prognostic markers and therapeutic targets. Int J Cancer 10: 157–166.
Wang Y, Huang S, Sah VP, Ross Jr J, Brown JH, Han J et al. (1998). Cardiac muscle cell hypertrophy and apoptosis induced by distinct members of the p38 mitogen-activated protein kinase family. J Biol Chem 273: 2161–2168.
Westermarck J, Li S, Jaakkola P, Kallunki T, Grenman R, Kähäri VM . (2000). Activation of fibroblast collagenase-1 expression by tumor cells of squamous cell carcinomas is mediated by p38 mitogen-activated protein kinase and c-Jun NH2-terminal kinase-2. Cancer Res 60: 7156–7162.
Westermarck J, Li SP, Kallunki T, Han J, Kähäri VM . (2001). p38 mitogen-activated protein kinase-dependent activation of protein phosphatases 1 and 2A inhibits MEK1 and MEK2 activity and collagenase-1 (MMP-1) gene expression. Mol Cell Biol 21: 2373–2383.
Wilkinson GW, Akrigg A . (1992). Constitutive and enhanced expression from the CMV major IE promoter in a defective adenovirus vector. Nucleic Acids Res 20: 2233–2239.
Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME . (1995). Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 270: 1326–1331.
Acknowledgements
We acknowledge the expert technical assistance of Sari Pitkänen, Johanna Markola and Marjo Hakkarainen. This study was supported by grants from the Academy of Finland (projects 203421, 114405 and 8212695), Sigrid Jusélius Foundation, the Cancer Research Foundation of Finland, Turku University Central Hospital (project 13336) and by European Union Framework Programme 6 (LSHC-CT-2003-503297; CANCERDEGRADOME).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Junttila, M., Ala-aho, R., Jokilehto, T. et al. p38α and p38δ mitogen-activated protein kinase isoforms regulate invasion and growth of head and neck squamous carcinoma cells. Oncogene 26, 5267–5279 (2007). https://doi.org/10.1038/sj.onc.1210332
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.onc.1210332
Keywords
This article is cited by
-
Suppressing MTERF3 inhibits proliferation of human hepatocellular carcinoma via ROS-mediated p38 MAPK activation
Communications Biology (2024)
-
Inhibition of TGF-β signaling, invasion, and growth of cutaneous squamous cell carcinoma by PLX8394
Oncogene (2023)
-
GFAT1-linked TAB1 glutamylation sustains p38 MAPK activation and promotes lung cancer cell survival under glucose starvation
Cell Discovery (2022)
-
p38 Expression and Modulation of STAT3 Signaling in Oral Cancer
Pathology & Oncology Research (2020)
-
GAGE7B promotes tumor metastasis and growth via activating the p38δ/pMAPKAPK2/pHSP27 pathway in gastric cancer
Journal of Experimental & Clinical Cancer Research (2019)
