
Abstract
Aberrant activation of the Wnt signaling pathway has been reported during neoplastic progression in Barrett's esophagus (BE). However, mutations in APC and CTNNB1 genes were rarely observed. In this study, expression pattern of Wnt ligands, Frizzled receptors and APC, as well as the methylation status of the APC, SFRP1 and SFRP2 promoter genes were investigated in normal esophageal mucosa and in preneoplastic and neoplastic lesions of BE patients. Promoter methylation of APC was found in all BE samples and in 95% of esophageal adenocarcinomas (EAC). Full methylation of APC correlated with lack of expression. In EAC, nuclear translocation of β-catenin was observed regardless of the expression of APC. WNT2 expression was higher in dysplasia and EAC than in BE, with 20/26 (77%) of the EAC showing high expression of WNT2. SFRP1 methylation occurred in all BE samples and in 96% of EAC, while SFRP2 was methylated in 73% of the normal squamous esophageal mucosa samples. In conclusion, (1) alterations of key regulators of the Wnt signaling are frequent in the pathogenesis of BE; (2) the APC and SFRP1 genes are inactivated by promoter methylation in BE; (3) the WNT2 gene is upregulated along the progression from low-grade dysplasia to EAC.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 50 print issues and online access
$259.00 per year
only $5.18 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others
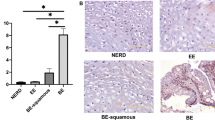

References
Baisse B, Bian YS, Benhattar J . (2000). Biotechniques 28: 856–858, 860, 862.
Benhattar J, Clement G . (2004). Methods Mol Biol 287: 181–193.
Bian YS, Osterheld MC, Bosman FT, Fontolliet C, Benhattar J . (2000). Am J Clin Pathol 114: 583–590.
Bian YS, Yan P, Osterheld MC, Fontolliet C, Benhattar J . (2001). Biotechniques 30: 66–72.
Bienz M, Clevers H . (2000). Cell 103: 311–320.
Blasband A, Schryver B, Papkoff J . (1992). Oncogene 7: 153–161.
Blot WJ, McLaughlin JK . (1999). Semin Oncol 26: 2–8.
Brabender J, Marjoram P, Salonga D, Metzger R, Schneider PM, Park JM et al. (2004). Oncogene 23: 4780–4788.
Bui TD, Zhang L, Rees MC, Bicknell R, Harris AL . (1997). Br J Cancer 75: 1131–1136.
Caldwell GM, Jones C, Gensberg K, Jan S, Hardy RG, Byrd P et al. (2004). Cancer Res 64: 883–888.
Clement G, Benhattar J . (2005). J Clin Pathol 58: 155–158.
Clement G, Bosman FT, Fontolliet C, Benhattar J . (2004). Cancer Res 64: 6867–6873.
Falk GW . (2002). Gastroenterology 122: 1569–1591.
Flejou JF . (2005). Gut 54: i6–i12.
Gonzalez MV, Artimez ML, Rodrigo L, Lopez-Larrea C, Menendez MJ, Alvarez V et al. (1997). J Clin Pathol 50: 212–217.
Guillou L, Coindre J, Gallagher G, Terrier P, Gebhard S, de Saint Aubain SN et al. (2001). Hum Pathol 32: 105–112.
He B, Lee AY, Dadfarmay S, You L, Xu Z, Reguart N et al. (2005). Cancer Res 65: 743–748.
Holcombe RF, Marsh JL, Waterman ML, Lin F, Milovanovic T, Truong T . (2002). Mol Pathol 55: 220–226.
Horii A, Nakatsuru S, Ichii S, Nagase H, Nakamura Y . (1993). Hum Mol Genet 2: 283–287.
Howng SL, Wu CH, Cheng TS, Sy WD, Lin PC, Wang C et al. (2002). Cancer Lett 183: 95–101.
Jenkins GJ, Doak SH, Parry JM, D'Souza FR, Griffiths AP, Baxter JN . (2002). Br J Surg 89: 824–837.
Katoh M . (2001a). Int J Mol Med 8: 657–660.
Katoh M . (2001b). Int J Oncol 19: 1003–1007.
Katoh M . (2003). Int J Mol Med 12: 811–816.
Kawano Y, Kypta R . (2003). J Cell Sci 116: 2627–2634.
Kitadai Y, Haruma K, Tokutomi T, Tanaka S, Sumii K, Carvalho M et al. (1998). Clin Cancer Res 4: 2195–2200.
Kobayashi K, Ouchida M, Tsuji T, Hanafusa H, Miyazaki M, Namba M et al. (2002). Gene 282: 151–158.
Koppert LB, van der Velden AW, van de Wetering M, Abbou M, van den Ouweland AM, Tilanus HW et al. (2004). Br J Cancer 90: 892–899.
Le Floch N, Rivat C, De WO, Bruyneel E, Mareel M, Dale T et al. (2005). FASEB J 19: 144–146.
Lee AY, He B, You L, Dadfarmay S, Xu Z, Mazieres J et al. (2004). Oncogene 23: 6672–6676.
Logan CY, Nusse R . (2004). Annu Rev Cell Dev Biol 20: 781–810.
McManus DT, Olaru A, Meltzer SJ . (2004). Cancer Res 64: 1561–1569.
Metzger R, Schneider PM, Warnecke-Eberz U, Brabender J, Holscher AH . (2004). Onkologie 27: 200–206.
Morin PJ . (1999). Bioessays 21: 1021–1030.
Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B et al. (1997). Science 275: 1787–1790.
Nishihira T, Hashimoto Y, Katayama M, Mori S, Kuroki T . (1993). J Cancer Res Clin Oncol 119: 441–449.
Nusse R, Varmus HE . (1982). Cell 31: 99–109.
Osterheld MC, Bian YS, Bosman FT, Benhattar J, Fontolliet C . (2002). Am J Clin Pathol 117: 451–456.
Pham K, Milovanovic T, Barr RJ, Truong T, Holcombe RF . (2003). Mol Pathol 56: 280–285.
Polakis P . (1999). Curr Opin Genet Dev 9: 15–21.
Polakis P . (2000). Genes Dev 14: 1837–1851.
Rhee CS, Sen M, Lu D, Wu C, Leoni L, Rubin J et al. (2002). Oncogene 21: 6598–6605.
Ricken A, Lochhead P, Kontogiannea M, Farookhi R . (2002). Endocrinology 143: 2741–2749.
Rockett JC, Larkin K, Darnton SJ, Morris AG, Matthews HR . (1997). Br J Cancer 75: 258–263.
Saitoh T, Mine T, Katoh M . (2002). Int J Mol Med 9: 515–519.
Seery JP, Syrigos KN, Karayiannakis AJ, Valizadeh A, Pignatelli M . (1999). Acta Oncol 38: 945–948.
Shih IM, Yu J, He TC, Vogelstein B, Kinzler KW . (2000). Cancer Res 60: 1671–1676.
Suzuki H, Watkins DN, Jair KW, Schuebel KE, Markowitz SD, Chen WD et al. (2004). Nat Genet 36: 417–422.
Tsuchiya T, Tamura G, Sato K, Endoh Y, Sakata K, Jin Z et al. (2000). Oncogene 19: 3642–3646.
Verma UN, Surabhi RM, Schmaltieg A, Becerra C, Gaynor RB . (2003). Clin Cancer Res 9: 1291–1300.
Worm J, Christensen C, Gronbaek K, Tulchinsky E, Guldberg P . (2004). Oncogene 23: 5215–5226.
Acknowledgements
We thank Patricia Martin, Maude Muriset, Adeline Cottier and Tian Lu for skillful laboratory work. This study was supported by a grant from the Ligue Suisse contre le Cancer (Grant KLS-01327-02-2003).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Clément, G., Braunschweig, R., Pasquier, N. et al. Alterations of the Wnt signaling pathway during the neoplastic progression of Barrett's esophagus. Oncogene 25, 3084–3092 (2006). https://doi.org/10.1038/sj.onc.1209338
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.onc.1209338
Keywords
This article is cited by
-
Zinc Gluconate Induces Potentially Cancer Chemopreventive Activity in Barrett’s Esophagus: A Phase 1 Pilot Study
Digestive Diseases and Sciences (2021)
-
The gut microbiome switches mutant p53 from tumour-suppressive to oncogenic
Nature (2020)
-
Targeting the immune milieu in gastrointestinal cancers
Journal of Gastroenterology (2020)
-
In-depth characterization of the Wnt-signaling/β-catenin pathway in an in vitro model of Barrett’s sequence
BMC Gastroenterology (2019)
-
Integrated molecular analysis reveals complex interactions between genomic and epigenomic alterations in esophageal adenocarcinomas
Scientific Reports (2017)
