
Abstract
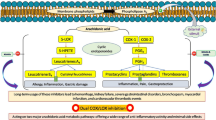
Nonsteroidal anti-inflammatory drugs (NSAIDs) such as aspirin have been shown to suppress transcription factor NF-κB, which controls the expression of genes such as cyclooxygenase (COX)-2 and cyclin D1, leading to inhibition of proliferation of tumor cells. There is no systematic study as to how these drugs differ in their ability to suppress NF-κB activation and NF-κB-regulated gene expression or cell proliferation. In the present study, we investigated the effect of almost a dozen different commonly used NSAIDs on tumor necrosis factor (TNF)-induced NF-κB activation and NF-κB-regulated gene products, and on cell proliferation. Dexamethasone, an anti-inflammatory steroid, was included for comparison with NSAIDs. As indicated by DNA binding, none of the drugs alone activated NF-κB. All compounds inhibited TNF-induced NF-κB activation, but with highly variable efficacy. The 50% inhibitory concentration required was 5.67, 3.49, 3.03, 1.25, 0.94, 0.60, 0.38, 0.084, 0.043, 0.027, 0.024, and 0.010 mM for aspirin, ibuprofen, sulindac, phenylbutazone, naproxen, indomethacin, diclofenac, resveratrol, curcumin, dexamethasone, celecoxib, and tamoxifen, respectively. All drugs inhibited IκBα kinase and suppressed IκBα degradation and NF-κB-regulated reporter gene expression. They also suppressed NF-κB-regulated COX-2 and cyclin D1 protein expression in a dose-dependent manner. All compounds inhibited the proliferation of tumor cells, with 50% inhibitory concentrations of 6.09, 1.12, 0.65, 0.49, 1.01, 0.19, 0.36, 0.012, 0.016, 0.047, 0.013, and 0.008 mM for aspirin, ibuprofen, sulindac, phenylbutazone, naproxen, indomethacin, diclofenac, resveratrol, curcumin, dexamethasone, celecoxib, and tamoxifen, respectively. Overall these results indicate that aspirin and ibuprofen are least potent, while resveratrol, curcumin, celecoxib, and tamoxifen are the most potent anti-inflammatory and antiproliferative agents of those we studied.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 50 print issues and online access
$259.00 per year
only $5.18 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout







Similar content being viewed by others

Abbreviations
- NSAIDs:
-
nonsteroidal anti-inflammatory drugs
- TNF:
-
tumor necrosis factor
- IκB:
-
inhibitory subunit of NF-κB
- IKK:
-
IκBα kinase
- EMSA:
-
electrophoretic mobility shift assays
- SEAP:
-
secretory alkaline phosphatase
- COX:
-
cyclooxygenase
References
Aggarwal BB . (2003). Nat. Rev. Immunol., 3, 745–756.
Ashikawa K, Majumdar S, Banerjee S, Bharti AC, Shishodia S and Aggarwal BB . (2002). J. Immunol., 169, 6490–6497.
Baron JA, Cole BF, Sandler RS, Haile RW, Ahnen D, Bresalier R, McKeown-Eyssen G, Summers RW, Rothstein R, Burke CA, Snover DC, Church TR, Allen JI, Beach M, Beck GJ, Bond JH, Byers T, Greenberg ER, Mandel JS, Marcon N, Mott LA, Pearson L, Saibil F and van Stolk RU . (2003). N. Engl. J. Med., 348, 891–899.
Bharti AC and Aggarwal BB . (2002). Biochem. Pharmacol., 64, 883–888.
Bharti AC, Takada Y, Shishodia S and Aggarwal BB . (2004). J. Biol. Chem., 279, 6065–6076.
Botting JH . (1999). Drugs Today (Barc.), 35, 225–235.
Bryant CE, Farnfield BA and Janicke HJ . (2003). Am. J. Vet. Res., 64, 211–215.
Callejas NA, Casado M, Bosca L and Martin-Sanz P . (2002). Hepatology, 35, 341–348.
Chuang SE, Yeh PY, Lu YS, Lai GM, Liao CM, Gao M and Cheng AL . (2002). Biochem. Pharmacol., 63, 1709–1716.
Coussens LM and Werb Z . (2002). Nature, 420, 860–867.
Ferlini C, Scambia G, Marone M, Distefano M, Gaggini C, Ferrandina G, Fattorossi A, Isola G, Benedetti Panici P and Mancuso S . (1999). Br. J. Cancer, 79, 257–263.
Fernandez de Arriba A, Cavalcanti F, Miralles A, Bayon Y, Alonso A, Merlos M, Garcia-Rafanell J and Forn J . (1999). Mol. Pharmacol., 55, 753–760.
Ghosh S and Karin M . (2002). Cell, 109 (Suppl), S81–S96.
Giercksky KE . (2001). Best Pract. Res. Clin. Gastroenterol., 15, 821–833.
Goel A, Chang DK, Ricciardiello L, Gasche C and Boland CR . (2003). Clin. Cancer Res., 9, 383–390.
Guttridge DC, Albanese C, Reuther JY, Pestell RG and Baldwin Jr AS . (1999). Mol. Cell. Biol., 19, 5785–5799.
Hass R, Brach M, Gunji H, Kharbanda S and Kufe D . (1992). Biochem. Pharmacol., 44, 1569–1576.
Hinz M, Krappmann D, Eichten A, Heder A, Scheidereit C and Strauss M . (1999). Mol. Cell. Biol., 19, 2690–2698.
Inoue H, Yokoyama C, Hara S, Tone Y and Tanabe T . (1995). J. Biol. Chem., 270, 24965–24971.
Jack DB . (1997). Lancet, 350, 437–439.
Jung M and Dritschilo A . (2001). Semin. Radiat. Oncol., 11, 346–351.
Kazmi SM, Plante RK, Visconti V, Taylor GR, Zhou L and Lau CY . (1995). J. Cell. Biochem., 57, 299–310.
Kisley LR, Barrett BS, Dwyer-Nield LD, Bauer AK, Thompson DC and Malkinson AM . (2002). Carcinogenesis, 23, 1653–1660.
Kopp E and Ghosh S . (1994). Science, 265, 956–959.
Lawrence T, Willoughby DA and Gilroy DW . (2002). Nat. Rev. Immunol., 2, 787–795.
Manna SK, Mukhopadhyay A and Aggarwal BB . (2000). J. Immunol., 164, 6509–6519.
Miller C, Zhang M, He Y, Zhao J, Pelletier JP, Martel-Pelletier J and Di Battista JA . (1998). J. Cell Biochem., 69, 392–413.
Moysich KB, Menezes RJ, Ronsani A, Swede H, Reid ME, Cummings KM, Falkner KL, Loewen GM and Bepler G . (2002). BMC Cancer, 2, 31.
Nasuhara Y, Adcock IM, Catley M, Barnes PJ and Newton R . (1999). J. Biol. Chem., 274, 19965–19972.
Niederberger E, Tegeder I, Vetter G, Schmidtko A, Schmidt H, Euchenhofer C, Brautigam L, Grosch S and Geisslinger G . (2001). FASEB J., 15, 1622–1624.
O'Neill EA . (1998). Nature, 396, 15–17.
Roth GJ and Calverley DC . (1994). Blood, 83, 885–898.
Sandler RS, Halabi S, Baron JA, Budinger S, Paskett E, Keresztes R, Petrelli N, Pipas JM, Karp DD, Loprinzi CL, Steinbach G and Schilsky R . (2003). N. Engl. J. Med., 348, 883–890.
Scheuren N, Bang H, Munster T, Brune K and Pahl A . (1998). Br. J. Pharmacol., 123, 645–652.
Singh S and Aggarwal BB . (1995). J. Biol. Chem., 270, 24995–25000.
Stark LA, Din FV, Zwacka RM and Dunlop MG . (2001). FASEB J., 15, 1273–1275.
Takada Y and Aggarwal BB . (2003a). J. Immunol., 171, 3278–3286.
Takada Y and Aggarwal BB . (2003b). J. Biol. Chem., 278, 23390–23397.
Takada Y, Khuri FR and Aggarwal BB . (2004). J. Biol. Chem., 279, 26287–26299.
Takada Y, Mukhopadhyay A, Kundu GC, Mahabeleshwar GH, Singh S and Aggarwal BB . (2003). J. Biol. Chem., 278, 24233–24241.
Tegeder I, Pfeilschifter J and Geisslinger G . (2001). FASEB J., 15, 2057–2072.
Vane JR . (1971). Nat. N. Biol., 231, 232–235.
Warner TD, Giuliano F, Vojnovic I, Bukasa A, Mitchell JA and Vane JR . (1999). Proc. Natl. Acad. Sci. USA, 96, 7563–7568.
Williams CS, Watson AJ, Sheng H, Helou R, Shao J and DuBois RN . (2000). Cancer Res., 60, 6045–6051.
Yamamoto K, Arakawa T, Ueda N and Yamamoto S . (1995). J. Biol. Chem., 270, 31315–31320.
Yamamoto Y, Yin MJ, Lin KM and Gaynor RB . (1999). J. Biol. Chem., 274, 27307–27314.
Yin MJ, Yamamoto Y and Gaynor RB . (1998). Nature, 396, 77–80.
Acknowledgements
We thank Mr Walter Pagel for carefully proof-reading the manuscript and providing valuable comments. Dr Aggarwal is a Ransom Horne Jr, Distinguished Professor of Cancer Research. This work was supported partially by the Clayton Foundation for Research (to BBA), Department of Defense US Army Breast Cancer Research Program Grant BC010610 (to BBA), a PO1 Grant (CA91844) from the National Institutes of Health on Lung Cancer Chemoprevention (to BBA), and a P50 Head and Neck SPORE Grant from the National Institutes of Health (to BBA).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Takada, Y., Bhardwaj, A., Potdar, P. et al. Nonsteroidal anti-inflammatory agents differ in their ability to suppress NF-κB activation, inhibition of expression of cyclooxygenase-2 and cyclin D1, and abrogation of tumor cell proliferation. Oncogene 23, 9247–9258 (2004). https://doi.org/10.1038/sj.onc.1208169
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.onc.1208169
Keywords
This article is cited by
-
Involvement of the tumour necrosis factor receptor system in glioblastoma cell death induced by palbociclib-heptamethine cyanine dye conjugate
Cell Communication and Signaling (2024)
-
Large-scale computational modelling of the M1 and M2 synovial macrophages in rheumatoid arthritis
npj Systems Biology and Applications (2024)
-
The role of miRNA and lncRNA in heterotopic ossification pathogenesis
Stem Cell Research & Therapy (2022)
-
The Intercellular Communication Between Mesenchymal Stromal Cells and Hematopoietic Stem Cells Critically Depends on NF-κB Signalling in the Mesenchymal Stromal Cells
Stem Cell Reviews and Reports (2022)
-
The Incidence and Severity of Post-ERCP Pancreatitis in Patients Receiving Standard Administration of NSAIDs: a Systematic Review and Meta-analysis
Journal of Gastrointestinal Surgery (2022)
