
Abstract
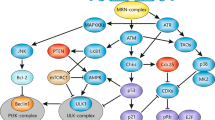
Damage induced in the DNA after exposure of cells to ionizing radiation activates checkpoint pathways that inhibit progression of cells through the G1 and G2 phases and induce a transient delay in the progression through S phase. Checkpoints together with repair and apoptosis are integrated in a circuitry that determines the ultimate response of a cell to DNA damage. Checkpoint activation typically requires sensors and mediators of DNA damage, signal transducers and effectors. Here, we review the current state of knowledge regarding mechanisms of checkpoint activation and proteins involved in the different steps of the process. Emphasis is placed on the role of ATM and ATR, as well on CHK1 and CHK2 kinases in checkpoint response. The roles of downstream effectors, such as P53 and the CDC25 family of proteins, are also described, and connections between repair and checkpoint activation are attempted. The role of checkpoints in genomic stability and the potential of improving the treatment of cancer by DNA damage inducing agents through checkpoint abrogation are also briefly outlined.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 50 print issues and online access
$259.00 per year
only $5.18 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others


References
Abraham RT . (2001). Genes Dev., 15, 2177–2196.
Agami R and Bernards R . (2000). Cell, 102, 55–66.
Amati B, Dalton S, Brooks MW, Littlewood TD, Evans GI and Land H . (1992). Nature, 359, 423–426.
Anderson CW and Carter TH . (1996). Curr. Top. Microbiol. Immunol., 217, 91–111.
Anderson L, Henderson C and Adachi Y . (2001). Mol. Cell. Biol., 21, 1719–1508.
Ando T, Kawabe T, Ohara H, Ducommun B and Itoh M . (2001). J. Biol. Chem., 276, 42971–42977.
Asaad NA, Zeng Z-C, Guan J, Thacker J and Iliakis G . (2000). Oncogene, 19, 5788–5800.
Atchley WR and Fitch WM . (1995). Proc. Natl. Acad. Sci. USA, 92, 10217–10221.
Bakkenist CJ and Kastan MB . (2003). Nature, 421, 499–506.
Banin S, Moyal L, Shieh S-Y, Taya Y, Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y and Ziv Y . (1998). Science, 281, 1674–1677.
Bao S, Tibbetts RS, Brumbaugh KM, Fang Y, Richardson DA, All A, Chen SM, Abraham RT and Wang X-F . (2001). Nature, 411, 969–974.
Bartek J, Falck J and Lukas J . (2001). Nat. Rev. Mol. Cell Biol., 2, 877–886.
Bartek J and Lukas C . (2001a). FEBS Lett., 490, 117–122.
Bartek J and Lukas J . (2001b). Curr. Opin. Cell Biol., 13, 738–747.
Bell SP and Dutta A . (2002). Annu. Rev. Biochem., 71, 333–374.
Bermudez VP, Lindsey-Boltz LA, Cesare AJ, Maniwa Y, Griffith JD, Hurwitz J and Sancar A . (2003). Proc. Natl. Acad. Sci. USA, 100, 1633–1638.
Bernhard EJ, Maity A, Muschel RJ and McKenna WG . (1995). Radiat. Environ. Biophys., 34, 79–83.
Bernhard EJ, McKenna WG and Muschel RJ . (1999). Cancer J., 5, 194–204.
Blasina A, Price BD, Turenne GA and McGowan CH . (1999). Curr. Biol., 9, 1135–1138.
Booher RN, Holman PS and Fattaey A . (1997). J. Biol. Chem., 272, 22300–22306.
Brown EJ and Baltimore D . (2000). Genes Dev., 14, 397–402.
Brush GS and Kelly TJ . (1996). Mechanisms for Replication DNA. Cold Spring Harbor Laboratory Press: New York.
Bulavin DV, Amundson SA and Fornace Jr AJ . (2002). Curr. Opin. Genet. Dev., 12, 92–97.
Canman CE, Lim D-S, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, Appella E, Kastan MB and Siliciano JD . (1998). Science, 281, 1677–1679.
Chan DW, Son S-C, Block W, Ye R, Khanna KK, Wold MS, Douglas P, Goodarzi AA, Pelley J, Taya Y, Lavin MF and Lees-Miller SP . (2000a). J. Biol. Chem., 275, 7803–7810.
Chan TA, Hermeking H, Lengauer C, Kinzler KW and Vogelstein B . (1999). Nature, 401, 616–620.
Chan TA, Hwang PM, Hermeking H, Kinzler KW and Vogelstein B . (2000b). Genes Dev., 14, 1584–1588.
Cleaver JE, Rose R and Mitchell DL . (1990). Radiat. Res., 124, 294–299.
Cliby WA, Roberts CJ, Cimprich KA, Stringer CM, Lamb JR, Schreiber SL and Friend SH . (1998). EMBO J., 17, 159–169.
Cortez D, Guntuku S, Qin J and Elledge SJ . (2001). Science, 294, 1713–1716.
Cortez D, Wang Y, Qin J and Elledge SJ . (1999). Science, 286, 1162–1166.
Costanzo V, Robertson K, Ying CY, Kim E, Avvedimento E, Gottesman M, Grieco D and Gautier J . (2000). Mol. Cell, 6, 649–659.
Crawford DF and Piwnica-Worms H . (2001). J. Biol. Chem., 276, 37166–37177.
D'Amours D, Desnoyers S, D'Silva I and Poirier GG . (1999). Biochem. J., 342, 249–268.
DePamphilis ML . (1999). BioEssays, 21, 5–16.
Desai-Mehta A, Cerosaletti KM and Concannon P . (2001). Mol. Cell. Biol., 21, 2184–2025.
DeSimone JN, Bengtsson U, Wang Q, Lao XY, Redpath JL and Stanbridge EJ . (2003). Radiat. Res., 159, 72–85.
DiBiase SJ, Zeng Z-C, Chen R, Hyslop T, Curran Jr WJ and Iliakis G . (2000). Cancer Res., 60, 1245–1253.
Donaldson AD and Blow JJ . (2001). Curr. Biol., 11, R979–R982.
Dotto GP . (2000). Biochim. Biophys. Acta, 1471, M43–M56.
Draetta G and Eckstein J . (1997). Biochim. Biophys. Acta, 1332, M53–M63.
Durocher D and Jackson SP . (2001). Curr. Opin. Cell Biol., 13, 225–231.
Dutta A and Bell SP . (1997). Ann. Rev. Cell Biol., 13, 293–332.
Edwards RJ, Bentley NJ and Carr AM . (1999). Nat. Cell Biol., 1, 393–398.
Eisenman RN and Cooper JA . (1995). Nature, 378, 438–439.
Elia AEH, Cantley LC and Yaffe MB . (2003). Science, 299, 1228–1231.
Elledge SJ . (1996). Science, 274, 1664–1672.
Falck J, Mailand N, Syljuasen RG, Bartek J and Lukas J . (2001). Nature, 410, 842–847.
Falck J, Petrini JHJ, Williams BR, Lukas J and Bartek J . (2002). Nat. Genet., 30, 290–294.
Friedberg EC, Walker GC and Siede W . (1995). DNA Repair Mutagenesis. ASM Press: Washington, DC.
Furnari B, Blasina A, Boddy MN, McGowan CH and Russell P . (1999). Mol. Biol. Cell, 10, 833–845.
Galaktionov K, Chen X and Beach D . (1996). Nature, 382, 511–517.
Gatei M, Scott SP, Filippovitch I, Soronika N, Lavin MF, Weber B and Khanna KK . (2000a). Cancer Res., 60, 3299–3304.
Gatei M, Young D, Cerosaletti KM, Desai-Mehta A, Spring K, Kozlov S, Lavin MF, Gatti RA, Concannon P and Khanna KK . (2000b). Nat. Genet., 25, 115–119.
Giaccia AJ and Kastan MB . (1998). Genes Dev., 12, 2973–2983.
Glover DM, Hagan IM and Tavares AAM . (1998). Genes Dev., 12, 3777–3787.
Goldberg M, Stucki M, Falck J, D'Amours D, Rahman D, Pappin D, Bartek J and Jackson SP . (2003). Nature, 421, 952–956.
Gottifredi V, Shieh SY, Taya Y and Prives C . (2001). Proc. Natl. Acad. Sci. USA, 98, 1036–1041.
Graves PR, Yu L, Schwarz JK, Gales J, Sausville EA, O'Connor PM and Piwnica-Worms H . (2000). J. Biol. Chem., 275, 5600–5605.
Green CM, Erdjument-Bromage H, Tempst P and Lowndes NF . (2000). Curr. Biol., 10, 39–42.
Griffiths DJ, Barbet NC, McCready S, Lehmann AR and Carr AM . (1995). EMBO J., 14, 5812–5823.
Guan J, DiBiase S and Iliakis G . (2000). Nucleic Acids Res., 28, 1183–1192.
Guo CY, D'Anna JA and Larner JM . (1999). Radiat. Res., 151, 125–132.
Guo Z, Kumagai A, Wang SX and Dunphy WG . (2000). Genes Dev., 14, 2745–2756.
Hagting A, Karlsson C, Clute P, Jackman M and Pines J . (1998). EMBO J., 17, 4127–4138.
Harper JW, Adami GR, Wei N, Keyomarsi K and Elledge SJ . (1993). Cell, 75, 805–816.
Harris EE, Kao GD, Muschel RJ and McKenna WG . (1998). Cancer Treat. Res., 93, 169–190.
Hartwell LH and Kastan MB . (1994). Science, 266, 1821–1828.
Hartwell LH and Weinert TA . (1989). Science, 246, 629–634.
Hermeking H, Lengauer C, Polyak K, He T-C, Zhang L, Thiagalingam S, Kinzler KW and Vogelstein B . (1997). Mol. Cell, 1, 3–11.
Hermeking H, Rago C, Schuhmacher M, Li Q, Barrett JF, Obaya AJ, O'Connell BC, Mateyak MK, Tam W, Kohlhuber F, Dang CV, Sedivy JM, Eick D, Vogelstein B and Kinzler KW . (2000). Proc. Natl. Acad. Sci. USA, 97, 2229–2234.
Hickman ES, Moroni MC and Helin K . (2002). Curr. Opin. Genet. Dev., 12, 60–66.
Hirao A, Kong YY, Matsuoka S, Wakeham A, Ruland J, Yoshida H, Liu D, Elledge SJ and Mak TW . (2000). Science, 287, 1824–1827.
Hoeijmakers JHJ . (2001). Nature, 411, 366–374.
Houldsworth J and Lavin MF . (1980). Nucleic Acids Res., 8, 3709–3720.
Hwang A, Maity A, McKenna WG and Muschel RJ . (1995). J. Biol. Chem., 270, 28419–28424.
Iliakis G . (1988). Int. J. Radiat. Biol., 53, 541–584.
Iliakis G . (1997). Semin. Oncol., 24, 602–615.
Jackson SP . (2002). Carcinogenesis, 23, 687–696.
Jeggo PA . (1997). Mutat. Res., 384, 1–14.
Jeggo PA . (1998). Adv. Genet., 38, 186–218.
Jin P, Gu Y and Morgan DO . (1996). J. Cell. Biol., 134, 963–970.
Jin P, Hardy S and Morgan DO . (1998). J. Cell Biol., 141, 875–885.
Kang D, Chen J, Wong J and Fang G . (2002). J. Cell Biol., 156, 249–259.
Kao GD, McKenna WG and Muschel RJ . (1999). J. Biol. Chem., 274, 34779–34784.
Kastan MB . (2001). Nature, 410, 766–767.
Kastan MB and Lim D-S . (2000). Nat. Rev. Mol. Cell Biol., 1, 179–186.
Kawabe T, Suganuma M, Ando T, Kimura M, Hori H and Okamoto T . (2002). Oncogene, 21, 1717–1726.
Khanna KK and Jackson SP . (2001). Nat. Genet., 27, 247–254.
Khanna KK, Keating KE, Kozlov S, Scott S, Gatei M, Hobson K, Taya Y, Gabrielli B, Chan D, Lees-Miller SP and Lavin MF . (1998). Nat. Genet., 20, 398–400.
Kim S-T, Lim D-S, Canman CE and Kastan MB . (1999). J. Biol. Chem., 274, 37538–37543.
Kim S-T, Xu B and Kastan MB . (2002). Genes Dev., 16, 560–570.
Lamb JR, Petit-Frere C, Broughton BC, Lehmann AR and Green MHL . (1989). Int. J. Radiat. Biol., 56, 125–130.
Larner JM, Lee H and Hamlin JL . (1997). Cancer Surv., 29, 25–45.
Larner JM, Lee H, Little RD, Dijkwel PA, Schildkraut CL and Hamlin JL . (1999). Nucleic Acids Res., 27, 803–809.
Larson JS, Tonkinson JL and Lai MT . (1997). Cancer Res., 57, 3351–3355.
Lavin MF and Schroeder AL . (1988). Mutat. Res., 193, 193–206.
Lee C-H and Chung JH . (2001). J. Biol. Chem., 276, 30537–30541.
Lee H, Larner JM and Hamlin JL . (1997). Proc. Natl. Acad. Sci. USA, 94, 526–531.
Lee J, Kumagai A and Dunphy WG . (2001). Mol. Biol. Cell, 12, 551–563.
Lee M and Nurse P . (1988). Trends Genet., 4, 287–290.
Lees-Miller SP . (1996). Biochem. Cell Biol., 74, 503–512.
Lehmann AR, Arlett CF, Burke JF, Green MHL, James MR and Lowe JE . (1986). Int. J. Radiat. Biol., 49, 639–643.
Leone G, DeGregori J, Sears R, Jakoi L and Nevins JR . (1997). Nature, 387, 422–425.
Li J, Meyer AN and Donoghue DJ . (1995). Mol. Biol. Cell, 6, 1111–1124.
Li S, Ting NSY, Zheng L, Chen P-L, Ziv Y, Shiloh Y, Lee EY-HP and Lee W-H . (2000). Nature, 406, 210–215.
Lim D-S, Kim S-T, Xu B, Maser RS, Lin J, Petrini JHJ and Kastan MB . (2000). Nature, 404, 613–617.
Lindsey-Boltz LA, Bernudez VP, Hurwitz J and Sancar A . (2001). Proc. Natl. Acad. Sci. USA, 98, 11236–11241.
Liu F-F, Stanton JJ, Wu Z and Piwnica-Worms H . (1997). Mol. Cell. Biol., 17, 571–583.
Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, Donehower LA and Elledge SJ . (2000). Genes Dev., 14, 1448–1459.
Lopez-Girona A, Kanoh J and Russell P . (2001). Curr. Biol., 11, 50–54.
Lou Z, Minter-Dykhouse K, Wu X and Chen J . (2003). Nature, 421, 957–961.
Lücke-Huhle C . (1982). Radiat. Res., 89, 298–308.
Lukas C, Bartkova J, Latella L, Falck J, Mailand N, Schroeder T, Sehested M, Lukas J and Bartek J . (2001). Cancer Res., 61, 4990–4993.
Lupardus PJ, Byun T, Yee M-c, Hekmat-Nejad M and Cimprich KA . (2002). Genes Dev., 16, 2327–2332.
Lydall D and Weinert T . (1995). Science, 270, 1488–1491.
Mailand N, Falck J, Lukas C, Syljuasen RG, Welcker M, Bartek J and Lukas J . (2000). Science, 288, 1425–1429.
Maity A, Hwang A, Janss A, Phillips P, McKenna WG and Muschel RJ . (1996). Oncogene, 13, 1647–1657.
Maity A, McKenna WG and Muschel RJ . (1994). Radiother. Oncol., 31, 1–13.
Marcu KB, Bossone SA and Patel AJ . (1992). Annu. Rev. Biochem., 61, 809–860.
Maser RS, Mirzoeva OK, Wells J, Olivares H, Williams BR, Zinkel RA, Farnham PJ and Petrini JHJ . (2001). Mol. Cell. Biol., 21, 6006–6016.
Matsuoka S, Huang M and Elledge SJ . (1998a). Science, 282, 1893–1897.
Matsuoka S, Huang M and Elledge SJ . (1998b). Science, 282, 1893–1897.
Matsuoka S, Rotman G, Ogawa A, Shiloh Y, Tamai K and Elledge SJ . (2000). Proc. Nat. Acad. Sci. USA, 97, 10389–10394.
McKenna WG . (1995). 37th Annual Meeting, American Society for Therapeutic Radiology and Oncology. Miami Beach, FL.
McKenna WG, Iliakis G, Weiss MC, Bernhard EJ and Muschel RJ . (1991). Radiat. Res., 125, 283–287.
Melchionna R, Chen X-B, Blasina A and McGowan CH . (2000). Nat. Cell Biol., 2, 762–765.
Metzger L and Iliakis G . (1991). Int. J. Radiat. Biol., 59, 1325–1339.
Morgan DO . (1995). Nature, 374, 131–134.
Muschel RJ, Zhang HB, Iliakis G and McKenna WG . (1992). Radiat. Res., 132, 153–157.
Nigg EA . (1998). Curr. Opin. Cell Biol., 10, 776–783.
Norbury C and Nurse P . (1992). Annu. Rev. Biochem., 61, 441–470.
Nurse P . (1990). Nature, 344, 503–508.
Nurse P . (1994). Cell, 79, 547–550.
Nurse P . (1997). Cell, 91, 865–867.
Nyberg KA, Michelson RJ, Putnam CW and Weinert TA . (2002). Annu. Rev. Genet., 36, 617–656.
O'Connell MJ, Walworth NC and Carr AM . (2002). Trends Cell Biol., 10, 296–303.
Paciotti V, Clerici M, Lucchini G and Longhese MP . (2000). Genes Dev., 14, 2046–2059.
Painter RB . (1981). Mutat. Res., 84, 183–190.
Painter RB . (1986). Int. J. Radiat. Biol., 49, 771–781.
Painter RB and Young BR . (1980). Proc. Nat. Acad. Sci. USA, 77, 7315–7317.
Parker AE, Van de Weyer I, Laus MC, Oostveen I, Yon J, Verhasselt P and Luyten WHML . (1998). J. Biol. Chem., 273, 18332–18339.
Parker LL and Piwnica-Worms H . (1992). Science, 257, 1955–1957.
Paulovich AG, Toczyski DP and Hartwell LH . (1997). Cell, 88, 315–321.
Peng C-Y, Graves PR, Thoma RS, Wu Z, Shaw AS and Piwnica-Worms H . (1997). Science, 277, 1501–1505.
Petrini JH . (2000). Curr. Opin. Cell Biol., 12, 293–296.
Pines J . (1995). Semin. Cancer Biol., 6, 63–72.
Pines J and Hunter T . (1991). J. Cell Biol., 115, 1–17.
Pines J and Hunter T . (1994). EMBO J., 13, 3772–3781.
Piwnica-Worms H . (1999). Nature, 401, 535–537.
Powell SN, DeFrank JS, Connell P, Eogan M, Preffer F, Dombkowski D, Tang W and Friend S . (1995). Cancer Res., 55, 1643–1648.
Rhind N and Russell P . (2000). J. Cell Sci., 113, 3889–3896.
Rotman G and Shiloh Y . (1998). Human Mol. Genet., 1998, 1555–1563.
Rotman G and Shiloh Y . (1999). Oncogene, 18, 6135–6144.
Rouse J and Jackson SP . (2000). EMBO J., 19, 5801–5812.
Sampath D and Plunkett W . (2001). Curr. Opin. Oncol., 13, 484–490.
Sanchez Y, Wong C, Thoma RS, Richman R, Wu Z, Piwnica-Worms H and Elledge SJ . (1997). Science, 277, 1497–1501.
Santoni-Rugiu E, Falck J, Mailand N, Bartek J and Lukas J . (2000). Mol. Cell. Biol., 20, 3497–3509.
Schultz LB, Chehab NH, Malikzay A and Halazonetis TD . (2000). J. Cell Biol., 151, 1381–1390.
Scolnick DM and Halazonetis TD . (2000). Nature, 406, 430–435.
Scully R and Livingston DM . (2000). Nature, 408, 429–442.
Senderowicz AM and Sausville EA . (2000). J. Nat. Cancer Inst., 92, 376–387.
Seoane J, Le H-V and Massague J . (2002). Nature, 419, 729–734.
Sheen J-H and Dickson RB . (2002). Mol. Cell. Biol., 22, 1819–1833.
Sherr CJ . (1995). Trends Biol. Sci., 20, 187–191.
Sherr CJ . (1996). Science, 274, 1672–1677.
Sherr CJ and Roberts JM . (1995). Genes Dev., 9, 1149–1163.
Sherr CJ and Roberts JM . (1999). Genes Dev., 13, 1501–1512.
Shiloh Y . (2001). Curr. Opin. Genet. Dev., 11, 71–77.
Sillje HHW and Nigg EA . (2003). Science, 299, 1190–1192.
Smeets MFMA, Mooren EHM, Abdel-Wahab AHA, Bartelink H and Begg AC . (1994). Radiat. Res., 140, 153–160.
Smith GCM, Cary RB, Lakin ND, Hann BC, Teo S-H, Chen DJ and Jackson SP . (1999). Proc. Natl. Acad. Sci. USA, 96, 11134–11139.
Smith GCM and Jackson SP . (1999). Genes Dev., 13, 916–934.
Smith S . (2001). Trends Biochem. Sci., 26, 175–180.
Smits VAJ, Klompmaker R, Arnaud L, Rijksen G, Nigg EA and Medema RH . (2000). Nat. Cell Biol., 2, 672–676.
Smits VAJ and Medema RH . (2001). Biochim. Biophys. Acta, 1519, 1–12.
Somasundaram K, Zhang H, Zeng YX, Houvras Y, Peng Y, Wu GS, Licht JD, Weber BL and El-Deiry WS . (1997). Nature, 389, 187–190.
Stewart GS, Wang B, Rignell CR, Taylor AMR and Elledge SJ . (2003). Nature, 421, 961–966.
Stillman B . (1994). Cell, 78, 725–728.
Stillman B . (1996). Science, 274, 1659–1664.
Stokes MP, Van Hatten R, Lindsay HD and Michael WM . (2002). J. Cell Biol., 158, 863–872.
Su L and Little JB . (1993). Radiat. Res., 133, 73–79.
Suganuma M, Kawabe T, Hori H, Funabiki T and Okamoto T . (1999). Cancer Res., 59, 5887–5891.
Takai H, Tominaga K, Motoyama N, Minamishima YA, Nagahama H, Tsukiyama T, Ikeda K, Nakayama K and Nakanishi M . (2000). Genes Dev., 14, 1439–1447.
Taylor WR, Agarwall ML, Agarwal A, Stacey D and Stark GR . (1999). Oncogene, 18, 283–295.
Taylor WR and Stark GR . (2001). Oncogene, 20, 1803–1815.
Terada Y, Tatsuka M, Jinno S and Okayama H . (1995). Nature, 376, 358–362.
Thompson LH and Schild D . (2001). Mutat. Res., 477, 131–153.
Thompson LH and Schild D . (2002). Mutat. Res., 509, 49–78.
Tibbetts RS, Brumbaugh KM, Williams JM, Sarkaria JN, Cliby WA, Shieh S-Y, Taya Y, Prives C and Abraham RT . (1999). Genes Dev., 13, 152–157.
Tibbetts RS, Cortez D, Brumbaugh KM, Scully R, Livingston D, Elledge SJ and Abraham RT . (2000). Genes Dev., 14, 2989–3002.
Tobey RA . (1975). Nature, 254, 245–247.
Toyoshima F, Moriguchi T, Wada A, Fukuda M and Nishida E . (1998). EMBO J., 17, 2728–2735.
Tye BK . (1999). Annu. Rev. Biochem., 68, 649–686.
van Gent DC, Hoeijmakers JHJ and Kanaar R . (2001). Nat. Rev. Genet., 2, 196–206.
van Vugt MATM, Smits VAJ, Klompmaker R and Medema RH . (2001). J. Biol. Chem., 276, 41656–41660.
Venkitaraman AR . (2001). J. Cell Sci., 114, 3591–3598.
Vogelstein B, Lane DP and Levine AJ . (2000). Nature, 408, 307–310.
Volkmer E and Karnitz LM . (1999). J. Biol. Chem., 274, 567–570.
Wakayama T, Kondo T, Ando S, Matsumoto K and Sugimoto K . (2001). Mol. Cell. Biol., 21, 755–764.
Walters RA, Gurley LR and Tobey RA . (1974). Biophys. J., 14, 99–118.
Walworth NC . (2001). Curr. Opin. Genet. Dev., 11, 78–82.
Wang H, Wang X, Iliakis G and Wang Y . (2003a). Radiat. Res., 159, 420–425.
Wang H, Zeng Z-C, Bui T-A, DiBiase SJ, Qin W, Xia F, Powell SN and Iliakis G . (2001a). Cancer Res., 61, 270–277.
Wang H, Zeng Z-C, Bui T-A, Sonoda E, Takata M, Takeda S and Iliakis G . (2001b). Oncogene, 20, 2212–2224.
Wang JYY . (2000). Nature, 405, 404–405.
Wang X, Wang H, Iliakis G and Wang Y . (2003b). Radiat. Res.. (in press).
Wang Y, Cortez D, Yazdi P, Neff N, Elledge SJ and Qin J . (2000). Genes Dev., 14, 927–939.
Wang Y, Huq MS, Cheng X and Iliakis G . (1995). Radiat. Res., 142, 169–175.
Wang Y, Zhou XY, Wang H-Y and Iliakis G . (1999). J. Biol. Chem., 274, 22060–22064.
Weichselbaum RR, Nove J and Little JB . (1978). Nature, 271, 261–262.
Wolkow TD and Enoch T . (2002). Mol. Biol. Cell, 13, 480–492.
Wu X, Ranganathan V, Weisman DS, Heine WF, Ciccone DN, O'Neill TB, Crick KE, Pierce KA, Lane WS, Rathbun G, Livingston DM and Weaver DT . (2000). Nature, 405, 477–482.
Xia F, Taghian DG, DeFrank JS, Zeng Z-C, Willers H, Iliakis G and Powell SN . (2001). Proc. Natl. Acad. Sci. USA, 98, 8644–8649.
Xie S, Wu H, Wang Q, Cogswell P, Husain I, Conn C, Stambrook P, Jhanwar-Uniyal M and Dai W . (2001). J. Biol. Chem., 276, 43305–43312.
Xiong Y, Zhang H and Beach D . (1992). Cell, 71, 505–514.
Xu B, Kim S-t and Kastan MB . (2001). Mol. Cell. Biol., 21, 3445–3450.
Xu B, Kim S-T, Lim D-S and Kastan MB . (2002). Mol. Cell. Biol., 22, 1049–1059.
Yamane K, Wu X and Chen J . (2002). Mol. Cell. Biol., 22, 555–566.
Yang J, Bardes ESG, Moore JD, Brennan J, Powers MA and Kornbluth S . (1998). Genes Dev., 12, 2131–2143.
Yarden RI, Pardo-Reoyo S, Sgagias M, Cowan KH and Brody LC . (2002). Nat. Genet., 30, 285–289.
Yazdi PT, Wang Y, Zhao S, Patel N, Lee EY-HP and Qin J . (2002). Genes Dev., 16, 571–582.
Yu Q, Geng Y and Sicinski P . (2001). Nature, 411, 1017–1021.
Zachos G, Rainey MD and Gillespie DAF . (2003). EMBO J., 22, 713–723.
Zhao H, Watkins JL and Piwnica-Worms H . (2002). Proc. Natl. Acad. Sci. USA, 99, 14795–14800.
Zhao S, Weng Y-C, Yuan S-SF, Lin Y-T, Hsu HC, Lin S-CJ, Gerbino E, Song M-h, Zdzienicka MZ, Gatti RA, Shay JW, Ziv Y, Shiloh Y and Lee EY-HP . (2000). Nature, 405, 473–477.
Zhou B-BS, Chaturvedi P, Spring K, Scott SP, Johanson RA, Mishra R, Mattern MR, Winkler JD and Khanna KK . (2000). J. Biol. Chem., 275, 10342–10348.
Zhou B-BS and Elledge SJ . (2000). Nature, 408, 433–439.
Zhou X-Y, Wang X, Hu B, Guan J, Iliakis G and Wang Y . (2002). Cancer Res., 62, 1598–1603.
Ziegler M and Oei SL . (2001). BioEssays, 23, 543–548.
Zou L, Cortez D and Elledge SJ . (2002). Genes Dev., 16, 198–208.
Acknowledgements
We are indebted to Jutta Mueller and Nancy Mott for secretarial assistance. The work in our is supported by NIH Grants CA42026, CA56706, CA76203 and 2P01 CA56690, EU Contracts FIS5-2002-00078 and FIGH-CT-2002-00218, as well as a Grant from the Volkswagenstiftung.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Iliakis, G., Wang, Y., Guan, J. et al. DNA damage checkpoint control in cells exposed to ionizing radiation. Oncogene 22, 5834–5847 (2003). https://doi.org/10.1038/sj.onc.1206682
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.onc.1206682
Keywords
This article is cited by
-
Role of the mechanical microenvironment on CD-44 expression of breast adenocarcinoma in response to radiotherapy
Scientific Reports (2024)
-
Chk1/2 inhibitor AZD7762 enhances the susceptibility of IDH-mutant brain cancer cells to temozolomide
Medical Oncology (2022)
-
Transcriptome profiling of cells exposed to particular and intense electromagnetic radiation emitted by the "SG-III" prototype laser facility
Scientific Reports (2021)
-
PM014 attenuates radiation-induced pulmonary fibrosis via regulating NF-kB and TGF-b1/NOX4 pathways
Scientific Reports (2020)
-
Radiation-dose-dependent functional synergisms between ATM, ATR and DNA-PKcs in checkpoint control and resection in G2-phase
Scientific Reports (2019)
