
Abstract
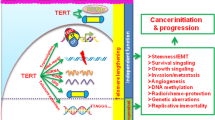
In the decade since the telomere hypothesis of cellular aging was proposed, the two essential genes for human telomerase were cloned and characterized, allowing experimental proof of the causal relationships between telomere loss and replicative senescence, and telomerase activation and immortalization. These relationships were established using a variety of cultured human cell types from both normal and tumor tissues, and were largely confirmed in the telomerase knockout mouse. Taken together, the data provide strong support for the potential utility of telomerase detection and inhibition for cancer, and telomerase activation for degenerative diseases. The specificity of the promoter for the telomerase catalytic gene and the antigenicity of the protein product, hTERT, provide additional strategies for killing telomerase-positive tumor cells. Unfortunately, the strong link between telomerase and cancer has led some to confuse telomerase activation with cancer, and others to overstate the cancer risk of telomerase activation therapies for degenerative diseases. This review clarifies the difference between telomerase, which does not cause growth deregulation, and oncogenes, which do. It also addresses the concept of telomerase repression as a tumor suppressor mechanism early in life, with detrimental tissue degeneration and tumor-promoting consequences late in life. This extended view of the telomere hypothesis helps explain how telomerase inhibition can be therapeutic in cancer patients, while controlled telomerase activation for degenerative diseases may actually reduce, rather than increase, the frequency of age-related tumorigenesis.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 50 print issues and online access
$259.00 per year
only $5.18 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others
References
Amit M, Carpenter MK, Inokuma MS, Chiu C-P, Harris CP, Waknitz MA, Itskovitz-Eldor J, Thomson JA . 2000 Dev. Biol. 227: 271–278
Bandyopadhyay D, Timchenko N, Suwa T, Hornsby PJ, Campisi J, Medrano EE . 2001 Exp. Gerontol. 36: 1265–1275
Blackburn EH . 2001 Cell 106: 661–673
Blackburn EH, Szostak JW . 1989 Ann. Rev. Genet. 23: 163–194
Bodnar A, Kim NW, Effros RB, Chiu C-P . 1996 Exp. Cell Res. 228: 58–64
Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu C-P, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE . 1998 Science 279: 349–352
Bryan TM, Marusic L, Bacchetti S, Namba M, Reddel RR . 1997 Hum. Mol. Gen. 6: 1–16
Campisi J . 1997 Eur. J. Cancer 33: 703–709
Chen J-L, Blasco MA, Greider CW . 2000 Cell 100: 503–514
Choi D, Whittier PS, Oshima J, Funk WD . 2001 FASEB J. 15: 1014–1020
Cong YS, Bachetti S . 2000 J. Biol. Chem. 275: 35665–35668
Dessain SK, Yu H, Reddel RR, Beijersbergen RL, Weinberg RA . 2000 Cancer Res. 60: 537–541
Dickson MA, Hahn WC, Ino Y, Ronfard V, Wu JY, Weinberg RA, Louis DN, Li FP, Rheinwald JG . 2000 Mol. Cell. Biol. 20: 1436–1447
Effros RB, Allsopp R, Chiu C-P, Hausner MA, Hirji K, Wang L, Harley CB, Villeponteau B, West MD, Giorgi JV . 1996 AIDS 10: F17–F22
Engelhardt M, Mackenzie K, Drullinsky P, Silver RT, Moore MA . 2000 Cancer Res. 60: 610–617
Farwell DG, Shera KA, Koop JI, Bonnet GA, Matthews CP, Reuther GW, Coltrera MD, McDougall JK, Klingelhutz AJ . 2000 Am. J. Pathol. 156: 1537–1547
Feng J, Funk WD, Wang S-S, Weinrich SL, Avilion AA, Chiu C-P, Adams RR, Chang E, Yu J, Le S, West MD, Harley CB, Andrews WH, Greider CW, Villeponteau B . 1995 Science 269: 1236–1241
Franco S, MacKenzie KL, Dias S, Alvarez S, Rafii S, Moore MA . 2001 Exp. Cell Res. 268: 14–25
Funk WD, Wang CK, Shelton DN, Harley CB, Pagon GD, Hoeffler WK . 2000 Exp. Cell. Res. 258: 270–278
Gonzalez-Suarez E, Samper E, Flores JM, Blasco MA . 2000 Nat. Genet. 26: 114–117
Goytisolo FA, Samper E, Martin-Caballero J, Finnon P, Herrera E, Flores JM, Bouffler SD, Blasco MA . 2000 J. Exp. Med. 192: 1625–1636
Greider CW . 1996 Annu. Rev. Biochem. 65: 337–365
Griffith JD, Comeau L, Rosenfield S, Stansel RM, Bianchi A, Moss H, de Lange T . 1999 Cell 97: 503–514
Hahn WC, Stewart SA, Brooks MW, York SG, Eaton E, Kurachi A, Beijersbergen RL, Knoll JHM, Meyerson M, Weinberg RA . 1999 Nat. Med. 5: 1164–1170
Halvorsen RL, Leibowitz G, Levine F . 1999 Mol. Cell. Biol. 19: 1864–1870
Halvorsen TL, Beattie GM, Lopez AD, Hayek A, Levine F . 2000 J. Endrocrinol. 166: 103–109
Harley CB . 1991 Mut. Res. 256: 271–282
Harley CB, Futcher AB, Greider CW . 1990 Nature 345: 458–460
Harley CB, Kim NW, Prowse KR, Weinrich SL, Hirsch KS, West MD, Bacchetti S, Hirte HW, Counter CM, Greider CW, Wright WE, Shay JW . 1994 Cold Spring Harbor Symp. Quant. Biol. 59: 307–315
Hemann M, Strong M, Hao L, Greider C . 2001 Cell 107: 67–77
Herrera E, Samper E, Martin-Caballero J, Flores JM, Lee H-W, Blasco MA . 1999 EMBO J. 18: 2950–2960
Hiyama E, Hiyama K, Yokoyama T, Mitsuura Y, Piatyszek MA, Shay JW . 1995a Nature Med. 1: 249–255
Hiyama K, Hirai Y, Kyoizumi S, Akiyama M, Hiyamas E, Piatyszek MA, Shay JW, Ishioka S, Yamakido M . 1995b J. Immunol. 155: 3711–3715
Hooijberg E, Ruizendaal JJ, Snijders PJF, Kueter EWM, Walboomers JMM, Spits H . 2000 J. Immunol. 165: 4239–4245
Jiang X-R, Jimenez G, Chang E, Frolkis M, Kusler B, Sage M, Beeche M, Bodnar AG, Wahl GM, Tlsty TD, Chiu C-P . 1999 Nat. Genet. 21: 111–114
Jones CJ, Kipling D, Morris M, Hepburn P, Skinner J, Bounacer A, Wyllie FS, Ivan M, Bartek J, Wynford-Thomas D, Bond JA . 2000 Mol. Cell. Biol. 20: 5690–5699
Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PLC, Coviello GM, Wright WE, Weinrich SL, Shay JW . 1994 Science 266: 2011–2014
Kiyono T, Foster SA, Koop JI, McDougall JK, Galloway DA, Klingelhutz AJ . 1998 Nature 396: 84–88
Komata T, Kondo Y, Kanzawa T, Hirohata S, Koga S, Sumiyohi H, Srinivasula S, Barna B, Germano I, Takakura M, Inoue M, Alnemri E, Shay JW, Kyo S, Kondo S . 2001 Cancer Res. 61: 5796–5802
Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J . 2001 Proc. Natl. Acad. Sci. USA 98: 12072–12077
Lee H-W, Blasco MA, Gottlieb G, Horner II JW, Greider CW, DePinho RA . 1998 Nature 392: 569–574
MacKenzie KL, Franco S, May C, Sadelain M, Moore MAS . 2000 Exp. Cell. Res. 259: 336–350
Majumdar AS, Hughes DE, Lichtsteiner SP, Wang Z, Lebkowski JS, Vasserot AP . 2001 Gene Ther. 8: 568–578
Matsunaga H, Handa JT, Aotaki-Keen A, Sherwood SW, West MD, Hjelmeland LM . 1999 Invest. Ophtalmol. Vis. Sci. 40: 197–202
McSharry BP, Jones CJ, Skinner JW, Kipling D, Wilkinson GWG . 2001 J. Gen. Virol. 82: 855–863
Meyerson M, Counter CM, Eaton EN, Ellisen LW, Steiner P, Caddle SD, Ziaugra L, Beijersbergen RL, Davidoff MJ, Liu Q, Bacchetti S, Haber DA, Weinberg RA . 1997 Cell 90: 785–795
Migliaccio M, Amacker M, Just T, Reichenbach P, Valmori D, Cerottini J-C, Romero P, Nabholz M . 2000 J. Immunol. 165: 4978–4984
Minev B, Hipp J, Firat H, Schmidt H, Langlade-Demoyen P, Zanetti M . 2000 Proc. Natl. Acad. Sci. USA 97: 4796–4801
Mitchell JR, Collins K . 2000 Mol. Cell. 6: 361–371
Nair SK, Hiser A, Boczkowski D, Majumdar A, Naoe M, Lebkowski J, Vieweg J, Gilboa E . 2000 Nat. Med. 6: 1011–1017
Nakamura TM, Morin GB, Chapman KB, Weinrich SL, Andrews WH, Lingner J, Harley CB, Cech TR . 1997 Science 277: 955–959
O'Hare MJ, Bond J, Clarke C, Takeuchi Y, Atherton AJ, Berry C, Moody J, Silver ARJ, Davies DC, Alsop AE, Neville AM, Jat PS . 2001 PNAS 98: 646–651
Ouellette MM, McDaniel LD, Wright WE, Shay JW, Schultz RA . 2000 Hum. Mol. Gen. 9: 403–411
Poole JC, Andrews LG, Tollefsbol TO . 2001 Gene 269: 1–12
Ramirez RD, Morales CP, Herbert B-S, Rohde J, Passons C, Shay JW, Wright WE . 2001 Genes Dev. 15: 398–403
Ranganathan V, Heine WF, Ciccone DN, Rudolph KL, Wu X, Chang S, Hai H, Ahearn IM, Livingston DM, Resnick I, Rosen F, Seemanova E, Jarolim P, DePinho RA, Weaver DT . 2001 Curr. Biol. 11: 962–966
Reynolds CP, Zuo JJ, Kim NW, Wang H, Lukens J, Matthay KK, Seeger RC . 1997 Eur. J. Cancer 33: 1929–1931
Rudolph KL, Chang S, Lee H-W, Blasco M, Goettlieb GJ, Greider C, DePinho RA . 1999a Cell 96: 701–712
Rudolph KL, Chang S, Millard M, Schreiber-Agus N, DePinho RA . 1999b Science 287: 1253–1258
Rudolph KL, Millard M, Bosenberg MW, DePinho RA . 2001 Nat. Genet. 28: 155–159
Salmon P, Oberholzer J, Occhiodoro T, Morel P, Lou J, Trono D . 2000 Mol. Ther. 2: 404–414
Seigneurin-Venin S, Bernard V, Ouellette MM, Mouly V, Wright WE, Tremblay J . 2000a Biochem. Biophys. Res. Commun. 7: 362–369
Seigneurin-Venin S, Bernard V, Tremblay JP . 2000b Gene Thera. 7: 619–623
Smith S, de Lange T . 2000 Curr. Biol. 10: 1299–1302
Smogorzewska A, van Steensel B, Bianchi A, Oelmann S, Schaefer MR, Schnapp G, de Lange T . 2000 Mol. Cell. Biol. 20: 1659–1668
Taylor RS, Ramirez RD, Ogoshi M, Chaffins M, Piatyszek MA, Shay JW . 1996 J. Invest. Dermatol. 106: 759–765
Thomas M, Yang L, Hornsby PJ . 2000 Nat. Biotech. 18: 39–42
Ulaner GA, Giudice LC . 1997 Mol. Hum. Reprod. 3: 769–773
Ulaner GA, Hu J-F, Hu TH, Giudice LC, Hoffman AR . 1998 Cancer Res. 58: 4168–4172
Usselmann B, Newbold M, Morris AG, Nwokolo CU . 2001 Am. J. Gastroenterol. 96: 1106–1112
Vaziri H, Benchimol S . 1999 Oncogene 18: 7676–7680
von Zglinicki T . 2000 Ann. NY Acad. Sci. 99–110
Vonderheide RH, Hahn WC, Schultze JL, Nadler LM . 1999 Immunity 10: 673–679
Vulliamy T, Marrone A, Goldman F, Dearlove A, Bessler M, Mason PJ, Dokal I . 2001 Nature 413: 432–435
Weng N-P, Granger L, Hodes RJ . 1997 Proc. Natl. Acad. Sci. USA 94: 10827–10832
Weng N-P, Levine BL, June CH, Hodes RJ . 1996 J. Exp. Med. 183: 2471–2479
Wong K-K, Chang S, Weiler SR, Ganesan S, Chaudhuri J, Zhu C, Artandi SE, Rudolph KL, Gottlieb GJ, Chin L, Alt FW, DePinho RA . 2000 Nat. Gen. 26: 85–88
Wood LD, Halvorsen TL, Dhar S, Baur JA, Pandita RK, Wright WE, Hande MP, Calaf G, Hei TK, Levine F, Shay JW, Wang JJ, Pandita TK . 2001 Oncogene 20: 278–288
Wright WE, Piatyszek MA, Rainey WE, Byrd W, Shay JW . 1996 Dev. Genet. 18: 173–179
Wright WE, Shay JW . 2001 Curr. Opin. Genet. Dev. 11: 98–103
Wyllie FS, Jones CJ, Skinner JW, Haughton MF, Wallis C, Wynford-Thomas D, Faragher RGA, Kipling D . 2000 Nat. Genet. 24: 16–17
Yang J, Chang E, Cherry AM, Bangs CD, Oei Y, Bodnar A, Bronstein A, Chiu C-P, Herron GS . 1999 J. Biol. Chem. 274: 26141–26148
Yudoh K, Matsuno H, Nakazawa F, Katayama R, Kimura T . 2001 J. Bone Miner. Res. 16: 1453–1464
Zhang X, Mar V, Harrington L, Robinson MO . 1999 Genes Dev. 13: 2388–2399
Acknowledgements
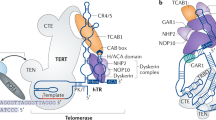
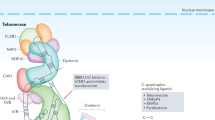
I thank my Geron colleagues for helpful discussion, and David Karpf, Tom Okarma and Jane Lebkowski for critical comments on this paper. I also thank Gregg Morin and Melissa Fischer for creation of the telomerase schematic upon which Figure 1 was based.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Harley, C. Telomerase is not an oncogene. Oncogene 21, 494–502 (2002). https://doi.org/10.1038/sj.onc.1205076
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.onc.1205076
Keywords
This article is cited by
-
Potent telomerase activators from a novel sapogenin via biotransformation utilizing Camarosporium laburnicola, an endophytic fungus
Microbial Cell Factories (2023)
-
The other side of the coin: mesenchymal stromal cell immortalization beyond evasion of senescence
Human Cell (2023)
-
Immortalization of Mesenchymal Stromal Cells by TERT Affects Adenosine Metabolism and Impairs their Immunosuppressive Capacity
Stem Cell Reviews and Reports (2020)
-
Implications of TERT promoter mutations and telomerase activity in urothelial carcinogenesis
Nature Reviews Urology (2018)
-
Design and Development of Artificial Zinc Finger Transcription Factors and Zinc Finger Nucleases to the hTERT Locus
Molecular Therapy - Nucleic Acids (2013)
