
Abstract
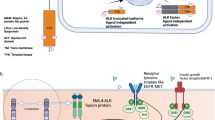
Anaplastic large-cell lymphoma (ALCL) comprises a group of non-Hodgkin's lymphomas (NHLs) that were first described in 1985 by Stein and co-workers and are characterized by the expression of the CD30/Ki-1 antigen (Stein et al., 1985). Approximately half of these lymphomas are associated with a typical chromosomal translocation, t(2;5)(p23;q35). Much confusion about the exact classification and clinicopathological features of this subgroup of NHL was clarified with the identification of NPM–ALK (nucleophosmin-anaplastic lymphoma kinase) as the oncogene created by the t(2;5) (Morris et al., 1994). With the discovery of NPM–ALK as the specific lymphoma gene mutation, this NHL subtype could be redefined on the molecular level. This achievement was enhanced by the availability of specific antibodies that recognize ALK fusion proteins in paraffin-embedded lymphoma tissues. Several excellent recent reviews have summarized the histopathological and molecular findings of ALCL and their use in the classification of this lymphoma entity (Anagnostopoulos and Stein, 2000; Benharroch et al., 1998; Drexler et al., 2000; Foss et al., 2000; Gogusev and Nezelof, 1998; Kadin and Morris, 1998; Ladanyi, 1997; Morris et al., 2001; Shiota and Mori, 1996; Skinnider et al., 1999; Stein et al., 2000). This review will focus on the molecular function and signal transduction pathways activated by ALK fusion oncogenes, with recent advances and possible clinical implications to be discussed.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 50 print issues and online access
$259.00 per year
only $5.18 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others

References
Anagnostopoulos I, Stein H . 2000 Pathology 21: 178–189
Bai RY, Coutinho S, Morris SW, Peschel C, Duyster J . 1998a Blood 92: 2110a
Bai RY, Dieter P, Peschel C, Morris SW, Duyster J . 1998b Mol. Cell. Biol. 18: 6951–6961
Bai RY, Ouyang T, Miething C, Morris SW, Peschel C, Duyster J . 2000 Blood 96: 4319–4327
Barbacid M . 1995 Ann. NY Acad. Sci. 766: 442–458
Beg AA, Baltimore D . 1996 Science 274: 782–784
Benharroch D, Meguerian-Bedoyan Z, Lamant L, Amin C, Brugieres L, Terrier-Lacombe MJ, Haralambieva E, Pulford K, Pileri S, Morris SW, Mason DY, Delsol G . 1998 Blood 91: 2076–2084
Beylot-Barry M, Groppi A, Vergier B, Pulford K, Merlio JP . 1998 Blood 91: 4668–4676
Bischof D, Pulford K, Mason DY, Morris SW . 1997 Mol. Cell. Biol. 17: 2312–2325
Borer RA, Lehner CF, Eppenberger HM, Nigg EA . 1989 Cell 56: 379–390
Brunel V, Sainty D, Carbuccia N, Arnoulet C, Costello R, Mozziconacci MJ, Simonetti J, Coignet L, Gabert J, Stoppa AM, Birg F, Lafagepochitaloff M . 1995 Genes Chrom. Cancer 14: 307–312
Butti MG, Bongarzone I, Ferraresi G, Mondellini P, Borrello MG, Pierotti MA . 1995 Genomics 28: 15–24
Cantley LC, Auger KR, Carpenter C, Duckworth B, Graziani A, Kapeller R, Soltoff S . 1991 Cell 64: 281–302
Chan PK, Chan FY . 1995 Biochim. Biophys. Acta 1262: 37–42
Chan PK Chan FY, Morris SW, Xie Z . 1997 Nucleic Acids Res. 25: 1225–1232
Chan PK, Liu QR, Durban E . 1990 Biochem. J. 270: 549–552
Chan WY, Liu QR, Borjigin J, Busch H, Rennert OM, Tease LA, Chan PK . 1989 Biochemistry 28: 1033–1039
Chauhan AK, Li YS, Deuel TF . 1993 Proc. Natl. Acad. Sci. USA 90: 679–682
Choudhuri R, Zhang HT, Donnini S, Ziche M, Bicknell R . 1997 Cancer Res. 57: 1814–1819
Colleoni GW, Bridge JA, Garicochea B, Liu J, Filippa DA, Ladanyi M . 2000 Am. J. Pathol. 156: 781–789
Cordell JL, Pulford KA, Bigerna B, Roncador G, Banham A, Colombo E, Pelicci PG, Mason DY, Falini B . 1999 Blood 93: 632–642
Courty J, Dauchel MC, Caruelle D, Perderiset M, Barritault D . 1991 Biochem. Biophys. Res. Commun. 180: 145–151
Czubayko F, Riegel AT, Wellstein A . 1994 J. Biol. Chem. 269: 21358–21363
Czubayko F, Schulte AM, Berchem GJ, Wellstein A . 1996 Proc. Natl. Acad. Sci. USA 93: 14753–14758
Delsol G, Lamant L, Mariame B, Pulford K, Dastugue N, Brousset P, Rigal-Huguet F, al Saati T, Cerretti DP, Morris SW, Mason DY . 1997 Blood 89: 1483–1490
Drexler HG, Gignac SM, von Wasielewski R, Werner M, Dirks WG . 2000 Leukemia 14: 1533–1559
Durkop H, Latza U, Hummel M, Eitelbach F, Seed B, Stein H . 1992 Cell 68: 421–427
Ellis TM, Simms PE, Slivnick DJ, Jack HM, Fisher RI . 1993 J. Immunol. 151: 2380–2389
Falini B, Bigerna B, Fizzotti M, Pulford K, Pileri SA, Delsol G, Carbone A, Paulli M, Magrini U, Menestrina F, Giardini R, Pilotti S, Mezzelani A, Ugolini B, Billi M, Pucciarini A, Pacini R, Pelicci PG, Flenghi L . 1998 Am. J. Pathol. 153: 875–886
Falini B, Pileri S, Zinzani PL, Carbone A, Zagonel V, Wolf-Peeters C, Verhoef G, Menestrina F, Todeschini G, Paulli M, Lazzarino M, Giardini R, Aiello A, Foss HD, Araujo I, Fizzotti M, Pelicci PG, Flenghi L, Martelli MF, Santucci A . 1999a Blood 93: 2697–2706
Falini B, Pulford K, Pucciarini A, Carbone A, De Wolf-Peeters C, Cordell J, Fizzotti M, Santucci A, Pelicci PG, Pileri S, Campo E, Ott G, Delsol G, Mason DY . 1999b Blood 94: 3509–3515
Fang W, Hartmann N, Chow DT, Riegel AT, Wellstein A . 1992 J. Biol. Chem. 267: 25889–25897
Foss HD, Marafioti T, Stein H . 2000 Pathologe 21: 124–136
Fujimoto J, Shiota M, Iwahara T, Seki N, Satoh H, Mori S, Yamamoto T . 1996 Proc. Natl. Acad. Sci. USA 93: 4181–4186
Gascoyne RD, Aoun P, Wu D, Chhanabhai M, Skinnider BF, Greiner TC, Morris SW, Connors JM, Vose JM, Viswanatha DS, Coldman A, Weisenburger DD . 1999 Blood 93: 3913–3921
Gogusev J, Nezelof C . 1998 Hematol. Oncol. Clin. North. Am. 12: 445–463
Greco A, Mariani C, Miranda C, Lupas A, Pagliardini S, Pomati M, Pierotti MA . 1995 Mol. Cell. Biol. 15: 6118–6127
Griffin CA, Hawkins AL, Dvorak C, Henkle C, Ellingham T, Perlman EJ . 1999 Cancer Res. 59: 2776–2780
Harris NL, Jaffe ES, Stein H, Banks PM, Chan JK, Cleary ML, Delsol G, De Wolf-Peeters C, Falini B, Gatter KC . 1994 Blood 84: 1361–1392
Hernandez L, Pinyol M, Hernandez S, Bea S, Pulford K, Rosenwald A, Lamant L, Falini B, Ott G, Mason DY, Delsol G, Campo E . 1999 Blood 94: 3265–3268
Hsu H, Shu HB, Pan MG, Goeddel DV . 1996 Cell 84: 299–308
Hübinger G, Scheffrahn I, Muller E, Bai R, Duyster J, Morris SW, Schrezenmeier H, Bergmann L . 1999a Exp. Hematol. 27: 1796–1805
Hübinger G, Wehnis E, Maurer U, Morris SW, Bergmann L . 1999b Blood 94: 598a
Iwahara T, Fujimoto J, Wen D, Cupples R, Bucay N, Arakawa T, Mori S, Ratzkin B, Yamamoto T . 1997 Oncogene 14: 439–449
Jiang YP, Wang H, D'Eustachio P, Musacchio JM, Schlessinger J, Sap J . 1993 Mol. Cell. Biol. 13: 2942–2951
Kadin ME . 1997 Cancer Surv. 30: 77–86
Kadin ME, Morris SW . 1998 Leuk. Lymphoma 29: 249–256
Khwaja A . 1999 Nature 401: 33–34
Kinney MC, Kadin ME . 1999 Am. J. Clin. Pathol. 111: S56–S67
Kinney MC, Greer JP, Kadin ME, DeCouteau JF, Collins RD . 1996 Lab. Invest. 74: 114A
Kuefer MU, Look AT, Pulford K, Behm FG, Pattengale PK, Mason DY, Morris SW . 1997 Blood 90: 2901–2910
Ladanyi M . 1997 Cancer Surv. 30: 59–75
Ladanyi M, Cavalchire G . 1996a Diagn. Mol. Pathol. 5: 154–158
Ladanyi M, Cavalchire G . 1996b Genes Chromos. Cancer 15: 173–177
Lamant L, Dastugue N, Pulford K, Delsol G, Mariame B . 1999 Blood 93: 3088–3095
Lamant L, Pulford K, Bischof D, Morris SW, Mason DY, Delsol G, Mariame B . 2000 Am. J. Pathol. 156: 1711–1721
Lawrence B, Perez-Atayde A, Hibbard MK, Rubin BP, Dal Cin P, Pinkus JL, Pinkus GS, Xiao S, Yi ES, Fletcher CD, Fletcher JA . 2000 Am. J. Pathol. 157: 377–384
Lee SY, Park CG, Choi Y . 1996 J. Exp. Med. 183: 669–674
Leevers SJ, Vanhaesebroeck B, Waterfield MD . 1999 Curr. Opin. Cell. Biol. 11: 219–225
Li S, Gillessen S, Tomasson MH, Dranoff G, Gilliland DG, van Etten RA . 2001 Blood 97: 1442–1450
Li YS, Milner PG, Chauhan AK, Watson MA, Hoffman RM, Kodner CM, Milbrandt J, Deuel TF . 1990 Science 250: 1690–1694
Liu QR, Chan PK . 1991 Eur. J. Biochem. 200: 715–721
Liu ZG, Hsu H, Goeddel DV, Karin M . 1996 Cell 87: 565–576
Luthra R, Pugh WC, Waasdorp M, Morris W, Cabanillas F, Chan PK, Sarris AH . 1998 Hematopathol. Mol. Hematol. 11: 173–183
Ma Z, Cools J, Marynen P, Cui X, Siebert R, Gesk S, Schlegelberger B, Peeters B, De Wolf-Peeters C, Wlodarska I, Morris SW . 2000 Blood 95: 2144–2149
Maeda N, Nishiwaki T, Shintani T, Hamanaka H, Noda A . 1996 J. Biol. Chem. 271: 21446–21452
Mahajan R, Delphin C, Guan T, Gerace L, Melchior F . 1997 Cell 88: 97–107
Mason DY, Pulford KA, Bischof D, Kuefer MU, Butler LH, Lamant L, Delsol G, Morris SW . 1998 Cancer Res. 58: 1057–1062
Michaud N, Goldfarb DS . 1991 J. Cell. Biol. 112: 215–223
Miething C, Bai R, Morris SW, von Schilling C, Schmidt B, Peschel C, Duyster J . 2000 Blood 96: 93a
Mir SS, Richter BW, Duckett CS . 2000 Blood 96: 4307–4312
Mitev L, Christova S, Hadjiev E, Guenova M, Oucheva R, Valkov I, Manolova Y . 1998 Leuk. Lymphoma 28: 613–616
Morris SW, Kirstein MN, Valentine MB, Dittmer KG, Shapiro DN, Saltman DL, Look AT . 1994 Science 263: 1281–1284
Morris SW, Naeve C, Mathew P, James PL, Kirstein MN, Cui X, Witte DP . 1997 Oncogene 14: 2175–2188
Morris SW, Xue L, Ma Z, Kinney MC . 2001 Br. J. Haematol. 113: 275–279
Nakamura S, Shiota M, Nakagawa A, Yatabe Y, Kojima M, Motoori T, Suzuki R, Kagami Y, Ogura M, Morishima Y, Mizoguchi Y, Okamoto M, Seto M, Koshikawa T, Mori S, Suchi T . 1997 Am. J. Surg. Pathol. 21: 1420–1432
Neckameyer WS, Shibuya M, Hsu MT, Wang LH . 1986 Mol. Cell. Biol. 6: 1478–1486
Nieborowska-Skorska M, Slupianek A, Zhang Q, Raghunath PN, Xue L, Morris SW, Wasik M, Skorski T . 2000 Blood 96: 2017a
Ochs R, Lischwe M, O'Leary P, Busch H . 1983 Exp. Cell. Res. 146: 139–149
Okuda M, Horn HF, Tarapore P, Tokuyama Y, Smulian AG, Chan PK, Knudsen ES, Hofmann IA, Snyder JD, Bove KE, Sukasawa K . 2000 Cell 103: 127–140
Park JP, Curran MJ, Levy NB, Davis TH, Elliott JH, Mohandas TK . 1997 Cancer Genet. Cytogenet. 96: 118–122
Pawson T, Gish D . 1992 Cell 71: 359–362
Peter M, Nakagawa J, Doree M, Labbe JC, Nigg EA . 1990 Cell 60: 791–801
Pfeifer W, Levi E, Petrogiannis-Haliotis T, Lehmann L, Wang Z, Kadin ME . 1999 Am. J. Pathol. 155: 1353–1359
Pileri SA, Pulford K, Mori S, Mason DY, Sabattini E, Roncador G, Piccioli M, Ceccarelli C, Piccaluga PP, Santini D, Leone O, Stein H, Falini B . 1997 Am. J. Pathol. 150: 1207–1211
Pulford K, Falini B, Banham AH, Codrington D, Roberton H, Hatton C, Mason DY . 2000 Blood 96: 1605–1607
Pulford K, Falini B, Cordell J, Rosenwald A, Ott G, Muller-Hermelink HK, MacLennan KA, Lamant L, Carbone A, Campo E, Mason DY . 1999 Am. J. Pathol. 154: 1657–1663
Pulford K, Lamant L, Morris SW, Butler LH, Wood KM, Stroud D, Delsol G, Mason DY . 1997 Blood 89: 1394–1404
Raulo E, Chernousov MA, Carey DJ, Nolo R, Rauvala H . 1994 J. Biol. Chem. 269: 12999–13004
Redner RL, Rush EA, Faas S, Rudert WA, Corey SJ . 1996 Blood 87: 882–886
Rodrigues GA, Park M . 1993 Mol. Cell. Biol. 13: 6711–6722
Rosenwald A, Ott G, Pulford K, Katzenberger T, Kuhl J, Kalla J, Ott MM, Mason DY, Muller-Hermelink HK . 1999 Blood 94: 362–364
Sandlund JT, Pui CH, Roberts WM, Santana VM, Morris SW, Berard CW, Hutchison RE, Ribeiro RC, Mahmoud H, Crist WM . 1994a Blood 84: 2467–2471
Sandlund JT, Pui CH, Santana VM, Mahmoud H, Roberts WM, Morris S, Raimondi S, Ribeiro R, Crist WM, Lin JS . 1994b J. Clin. Oncol. 12: 895–898
Schulte AM, Lai S, Kurtz A, Czubayko F, Riegel AT, Wellstein A . 1996 Proc. Natl. Acad. Sci. USA 93: 14759–14764
Schulte AM, Wellstein A . 1997 Tumor Angiogenesis Bicknell R, Lewis CM and Ferrara N. (eds). Oxford University Press: Oxford, New York, Tokyo pp. 273–289
Schwab U, Stein H, Gerdes J, Lemke H, Kirchner H, Schaadt M, Diehl V . 1982 Nature 299: 65–67
Sexl V, Piekorz R, Moriggl R, Rohrer J, Brown MP, Bunting KD, Rothammer K, Roussel MF, Ihle JN . 2000 Blood 96: 2277–2283
Shiota M, Mori S . 1996 Leuk. Lymphoma 23: 25–32
Shiota M, Fujimoto J, Semba T, Satoh H, Yamamoto T, Mori S . 1994a Oncogene 9: 1567–1574
Shiota M, Fujimoto J, Takenaga M, Satoh H, Ichinohasama R, Abe M, Nakano M, Yamamoto T, Mori S . 1994b Blood 84: 3648–3652
Shiota M, Nakamura S, Ichinohasama R, Abe M, Akagi T, Takeshita M, Mori N, Fujimoto J, Miyauchi J, Mikata A, Nanba K, Takami T, Yamabe H, Takano Y, Izumo T, Nagatani T, Mohri N, Nasu K, Satoh H, Katano H, Fujimoto J, Yamamoto T, Mori S . 1995 Blood 86: 1954–1960
Skinnider BF, Connors JM, Sutcliffe SB, Gascoyne RD . 1999 Hematol. Oncol. 17: 137–148
Slupianek A, Nieborowska-Skorska M, Hoser G, Morrione A, Majewski M, Xue L, Morris SW, Wasik MA, Skorski T . 2001 Cancer Res. 61: 2194–2199
Smith CA, Gruss HJ, Davis T, Anderson D, Farrah T, Baker E, Sutherland GR, Brannan CI, Copeland NG, Jenkins NA . 1993 Cell 73: 1349–1360
Souttou B, Juhl H, Hackenbruck J, Rockseisen M, Klomp HJ, Raulais D, Vigny M, Wellstein A . 1998 J. Natl. Cancer Inst. 90: 1468–1473
Stein H, Foss HD, Durkop H, Marafioti T, Delsol G, Pulford K, Pileri S, Falini B . 2000 Blood 96: 3681–3695
Stein H, Mason DY, Gerdes J, O'Connor N, Wainscoat J, Pallesen G, Gatter K, Falini B, Delsol G, Lemke H, Schwarting R, Lennert K . 1985 Blood 66: 848–858
Stoica GE, Kuo A, Aigner A, Sunitha I, Souttou B, Malercyk C, Caughey DJ, Went D, Karavanov A, Riegel AT, Wellstein A . 2001 J. Biol. Chem. 18: 16772–16779
Szebeni A, Herrera JE, Olson MO . 1995 Biochemistry 34: 8037–8042
Tian ZG, Longo DL, Funakoshi S, Asai O, Ferris DK, Widmer M, Murphy WJ . 1995 Cancer Res. 55: 5335–5341
Touriol C, Greenland C, Lamant L, Pulford K, Bernard F, Rousset T, Mason DY, Delsol G . 2000 Blood 95: 3204–3207
Trinei M, Lanfrancone L, Campo E, Pulford K, Mason DY, Pelicci PG, Falini B . 2000 Cancer Res. 60: 793–798
Trumper L, Pfreundschuh M, Bonin FV, Daus H . 1998 Br. J. Haematol. 103: 1138–1144
Ueno H, Honda H, Nakamoto T, Yamagata T, Sasaki K, Miyagawa K, Mitani K, Yazaki Y, Hirai H . 1997 Oncogene 14: 3067–3072
Ueno H, Sasaki K, Kozutsumi H, Miyagawa K, Mitani K, Yazaki Y, Hirai H . 1996 J. Biol. Chem. 271: 27707–27714
Valdez BC, Perlaky L, Henning D, Saijo Y, Chan PK, Busch H . 1994 J. Biol. Chem. 269: 23776–23783
Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM . 1996 Science 274: 787–789
Wellstein A, Fang WJ, Khatri A, Lu Y, Swain SS, Dickson RB, Sasse J, Riegel AT, Lippman ME . 1992 J. Biol. Chem. 267: 2582–2587
Yoneda-Kato N, Look AT, Kirstein MN, Valentine MB, Raimondi SC, Cohen KJ, Carroll AJ, Morris SW . 1996 Oncogene 12: 265–275
Yung BY, Busch H, Chan PK . 1985 Biochim. Biophys. Acta 826: 167–173
Zatsepina OV, Rousselet A, Chan PK, Olson MO, Jordan EG, Bornens M . 1999 J. Cell. Sci. 112: 455–466
Acknowledgements
This work was supported by a grant from the Wilhelm-Sander Stiftung and Mildred-Scheel Stiftung to J Duyster, by SFB grant No 456 to J Duyster, by National Cancer Institute (NCI) grants CA69129 and CA76301, and CORE grant CA21765 to SW Morris, and by the American-Lebanese Syrian Associated Charities (ALSAC), St Jude Children's Research Hospital.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Duyster, J., Bai, RY. & Morris, S. Translocations involving anaplastic lymphoma kinase (ALK). Oncogene 20, 5623–5637 (2001). https://doi.org/10.1038/sj.onc.1204594
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.onc.1204594
Keywords
This article is cited by
-
Novel TENM3–ALK fusion is an alternate mechanism for ALK activation in neuroblastoma
Oncogene (2022)
-
Prediction of Resistance Mutations Against Upcoming Anaplastic Lymphoma Kinase Inhibitors
Targeted Oncology (2022)
-
Recombinant expression, characterization, and quantification in human cancer cell lines of the Anaplastic Large-Cell Lymphoma-characteristic NPM-ALK fusion protein
Scientific Reports (2020)
-
Activation of IGF-1R pathway and NPM-ALK G1269A mutation confer resistance to crizotinib treatment in NPM-ALK positive lymphoma
Investigational New Drugs (2020)
-
KRCA-0008 suppresses ALK-positive anaplastic large-cell lymphoma growth
Investigational New Drugs (2020)
