
Abstract
Human X-linked dominant hypophosphatemic rickets (HPDR I) is characterized by hypophosphatemia, hyperphosphaturia, abnormal vitamin D metabolism, and rickets/osteomalacia. Two closely linked hypophosphatemic genes, hypophosphatemia (Hyp) and Gyro (Gy), are known on the mouse X chromosome. The Hyp phenotype is the equivalent of the human X-linked hypophosphatemia, while the human equivalent of the Gyro mouse has not been unambiguously identified. We observed an Italian four-generation pedigree with a new form of X-linked recessive hypophosphatemic rickets (XLRH). We demonstrated that HPDR I and XLRH are two different X-linked genes and that XLRH maps in the Xp11.2 region at 0% recombination fraction from the DXS1039 locus. We discuss this new finding in relation to the identification of the human equivalent of the Gyro mouse and to the recent mapping in Xp11.22 of another X-linked recessive renal disorder named Dent disease.
Similar content being viewed by others


Introduction
Rickets can be defined as a disorder in a growing person in which a lag in bone mineralization results in the accumulation of an abnormal amount of bone matrix (osteoid) in the tissue [1]. Since correct mineralization depends on adequate concentrations of extracellular calcium and phosphate, rickets may result from a dietary deficiency or gastrointestinal malabsorption of calcium, phosphate and/or vitamin D, from various abnormalities in vitamin D metabolism, and from renal phosphate tubulopathies as in the case of X-linked dominant (vitamin-D-resistant) hypophosphatemic rickets (HPDR I, XLH, HYP, McKusick No. 307800). HPDR I is the most common form of familial hypophosphatemia, characterized by a low serum level of phosphate, hyperphosphaturia with a decreased TmPO4/GFR (maximum tubular transport normalized to glomerular filtration rate), an inappropriately normal serum level of calcitriol (1,25-(OH)2D3; 1α,25-dihydroxycholecalciferol), rickets and/or osteomalacia.
Biochemical studies using the mouse model for human X-linked hypophosphatemia, the Hyp mouse, have localized the defect to the brush border membrane of the renal proximal convoluted tubule [2, 3]. However, it is still unknown whether this defect is a primary consequence of the disease gene or results from the abnormal function of a humoral factor which regulates renal phosphate transport. Thus, after the initial localization of the HPDR I gene between DXS43 and DXS41 in the Xp22.1–p22.2 region [4–8] and the subsequent refined genetic mapping [9], only the cloning of the gene may unravel the molecular basis of the disease.
More recently, Lyon et al. [10] identified a second X-linked hypophosphatemic mouse gene, namely Gy, which maps 1 cM distal to Hyp and 9 cM proximal to the Cream locus [11]. In addition to small stature, rickets, hypophosphatemia, and impaired renal phosphate reabsorption [10, 12], the Gyro mouse is characterized by inner ear abnormalities, circling behaviour, and hyperactivity [10]. With regard to the regulation of vitamin D metabolism, contrasting findings have been obtained by two different groups. Davidai et al. [13] have found that, unlike the Hyp mouse, the Gyro mouse shows normally regulated 1,25-(OH)2D3 synthesis because phosphate depletion, parathyroid hormone (PTH) stimulation, and calcitonin administration led to an increase of 1,25-(OH)2D3 serum level, gastrointestinal calcium absorption, and calciuria. On the other hand, according to Tenenhouse et al. [14], the Gyro mice exhibit a fall in plasma 1,25-(OH)2D3 and a rise in the renal vitamin D degradative pathway. Following the hypothesis that the X chromosome of all mammals carries homologous genes, it has been suggested that a second hypophosphatemic gene, homologous to Gy, might be present on the human X chromosome.
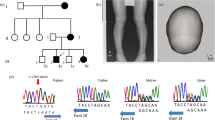
A number of patients with X-linked hypophosphatemia (XLH) and hearing impairment have been described by Boneh et al. [15], who suggested that they may represent the human counterpart of the Gyro mouse [15]. Enia et al. [16] observed an Italian four-generation family (fig. 1) with a new form of hypophosphatemic rickets. Transmission of the disease in this family is consistent with X-linked recessive inheritance and the proposed denomination is therefore X-linked recessive hypophosphatemia (XLRH). We performed a linkage study in the family described by Enia et al. [16] demonstrating that HPDR I and XLRH are caused by mutations at two different loci on the X chromosome. Our results indicate that the XLRH gene is excluded from the HPDR I region in Xp22.1–p22.2 and that the most likely localization of this new gene is in Xp11.2.

■ = Hypophosphatemia, hypercalciuria, high 1,25-(OH)2D3, rickets/osteomalacia; ■* = hypophosphatemia, hypercalciuria, high 1,25-(OH)2D3, rickets/osteomalacia plus inner ear defect; ▨,  = hypercalciuria alone. The haplotypes derived for the markers producing positive lod score values are also shown in this figure. No recombinants between the XLRH gene and DSX1039 were found.
= hypercalciuria alone. The haplotypes derived for the markers producing positive lod score values are also shown in this figure. No recombinants between the XLRH gene and DSX1039 were found.
Material and Methods
Phenotypes of Patients
Inheritance of hypophosphatemic rickets in the G.C. pedigree reported in figure 1 is consistent with X-linked recessive transmission. A total of 20 family members including the 5 affected males were analyzed for this linkage study. The clinical and biochemical characteristics of this family have been described by Enia et al. [16].
The five affected males have rickets/osteomalacia, hypophosphatemia with a decreased TmPO4/GFR, hypercalciuria, increased 1,25-(OH)2D3 serum levels, and progressive renal failure with nephrocalcinosis (table 1). Mild proteinuria was the first sign of the disease in the cases studied at an early stage. The normal family members 2, 3, 4, 9, 11, 14 and 15 (see fig. 1) have hypercalciuria alone with the other biochemical values in the normal range. Hypercalciuria in these individuals was considered as an independent trait which is very frequent in the general population (see discussion). Individual No. 17 (33 years old) has renal phosphate transport, phosphatemia, and a 1,25-(OH)2D3 serum level in the low-normal range. In order to define more precisely his phenotype, bone biopsies were carried out and a mild degree of osteomalacia was found. However, on the basis of the criteria listed above, this individual cannot be considered as affected because he shows only osteomalacia as a possible manifestation of the disease. However, since this finding does not allow us to consider individual No. 17 as normal, we excluded him from the linkage analysis.
Typing for Restriction Fragment Length Polymorphisms (RFLPs) and Short Tandem Repeat Polymorphisms (STRPs)
28 informative RFLP and STRP markers used in the linkage analysis are shown in table 2. DNA isolation, electrophoresis, Southern blotting, hybridization and autoradiography for the 21 RFLPs were performed using standard methods [17]. The BglI and BglII polymorphisms at the TIMP locus [18] and the DraI polymorphism at the OTC locus [19] were analyzed by PCR. Data from different RFLPs of the same locus were combined and considered as a single haplotype. In this way, the TIMP locus and the OATL1 locus were considered as 4-allele systems.
PCR amplifications for DXS458, DXS1055, DXS1039, DXS991, DXS1204 and DXS1000 were performed as described elsewhere [20, 21]. Microsatellite analysis was carried out following previously described conditions [22]. The primer sequences for DXS991 and DXS1000 are deposited in Genome Data Base, GDB. Those for DXS1055 were forward ATGGGATACACTGTTCTGGG and reverse TTAAACAATGCACAACTGGG; for DXS1039, forward CTCCTGTTCCTGGTATGTGA and reverse AGAAGAATGCCTGTTNGGGT, and for DXS1204, forward ATGAACCCTTAACTCATTTAGCAGG and reverse AGCNTGCACCAACATGCC. Analysis of the trinucleotide repeat corresponding to the AR locus was performed as described by Edwards et al. [23] with the following modifications: the amplification reaction was performed with unmodified oligonucleotides, and the cold amplification product was detected in 18% Polyacrylamide gel electrophoresis.
Linkage Analysis
Conventional two-point linkage analysis between the hypophosphatemic locus and each marker, and multipoint linkage analysis were carried out using the MLINK, ILINK and LINKMAP programs [24]. For the purpose of linkage analysis, XLRH was defined as an X-linked recessive fully penetrant disease, with a gene frequency of 0.001. Genetic mapping of DNA markers was performed as previously described [25].
Results
Exclusion of the XLRH Locus from the Xp22.1–p22.2 Region
To test whether HPDR I and XLRH correspond to two different genes on the X chromosome, we performed conventional two-point linkage analysis using 7 RFLPs from the Xp22.1–p22.2 region (table 2). Negative lod scores were obtained for all these loci (table 3).
Multipoint linkage analysis performed using 6 DNA markers from this region yielded multipoint lod scores of less than −2 for the whole region between DXS85 and DXS28 (fig. 2), including the interval between DXS274 and DXS92 where HPDR I has been previously mapped [26]. The genetic map used in the multipoint linkage analysis was derived from Alitalo et al. [27].

Multipoint linkage analysis with DNA markers from the HPDR I region. Recombination fractions were converted into centiMorgans using the Haldane mapping function.
Localization of the XLRH Locus
Two-point linkage analysis between XLRH and several DNA markers from various regions of the X chromosome (table 2) was performed. Negative lod scores were obtained for all markers (data not shown) except for those localized in the Xp11.22-p11.23 region (table 4). In particular, the RFLPs at the SYP, DXS146, and TFE3 loci showed no recombinations with XLRH. However, none of these was informative in all the meioses analyzed.
To increase the informativity in this candidate region, we performed the same analysis with microsatellite markers DXS1055, DXS1039, DXS1204, DXS991 and DXS1000 which have been recently identified [25]. As can be seen from the haplotype reconstruction of figure 1, no recombinants were observed between XLRH and the DXS1039 locus which showed a maximum lod score of 3.21 at 0% recombination fraction (1 lod unit support interval: 0–18.1%, table 4). The 1 lod unit support interval for the DXS1055 locus producing the second significant positive lod score of 2.23 at 6% recombination fraction is 0.3–31.2%.
Multipoint linkage analysis performed on a subset of the CEPH panel as already described [25], yielded the following most likely order and genetic distances (in cM) for three of the microsatellites and DXS7: tel — DXS7 — (16) — DXS1055 — (8.6) — DXS1039 — (3.8) — DXS991 — cen. Odds favoring this order against inversions of the microsatellites were at least 253:1. Odds against location of DXS7 in another interval were at least 5,970:1. The same three microsatellite markers have been physically assigned to the short arm of the X chromosome using three human-hamster somatic cell hybrids retaining Xp only, Xq only, and the whole X chromosome (data not shown).
A multipoint linkage analysis using the above genetic map localized the XLRH gene in the Xp11.2 region at a recombination fraction of 0% from the DXS1039 locus with a multipoint lod score of 3.82 (fig. 3).

Multipoint linkage analysis of the XLRH gene with Xp11.2 markers. A maximum multipoint lod score value of 3.82 has been obtained with the DXS1039 locus. Recombination fractions were converted into centiMorgans using the Haldane mapping function.
One lod unit support interval spans from 7.2% recombination fraction distal to 2.7% recombination fraction proximal to DXS1039. The odds favoring the location of XLRH at 0 distance from DXS1039 against the other possible locations in the multipoint map are at least 340:1.
Discussion
Ohno’s law [28] postulates that as a consequence of X chromosome inactivation, X-linked genes are conserved on the same chromosome in all mammalian species. A detailed and convincing demonstration of this hypothesis comes from the comparative mapping between humans and mouse. In these species, the X chromosomes share homology only between themselves and not with autosomes. The only known exception consists of the CSF2RA gene [29] which maps to human Xp22.32, and to mouse chromosome 19.
In particular, on the basis of the distribution of homologous loci on the human and mouse X chromosomes, it is possible to hypothesize that only a few rearrangements occurring during evolution are responsible for the different relative order of clusters of genes in the two species [30–32]. In most cases, this hypothesis can be used to predict the regional localization of human X-linked genes on the basis of the position of the corresponding loci on the mouse X chromosome, and vice versa.
Following this model, Buckle et al. [31] predicted that the human equivalent of the mouse hypophosphatemic locus Hyp may reside either between GLA (Xq21.3–q22) and HPRT (Xq26.1), or in the distal region of the short arm of the X chromosome. The last prediction was confirmed by Thakker et al. [6] who mapped the HPDR I gene by linkage analysis to the Xp22.1–p22.2 region between DXS43 and DXS41.
The identification in the mouse of a second X-linked locus responsible for a different form of hypophosphatemic rickets, namely Gy, suggested that an additional locus could also be present in humans.
Our Italian four-generation pedigree shows a new form of X-linked recessive hypophosphatemic rickets, XLRH [16]. This new disease shares with the classical X-linked hypophosphatemia (HPDR I) some important features like hypophosphatemia, reduced tubular reabsorption of phosphate, rickets and/or osteomalacia and, at the same time, shows the following peculiarities: (a) normal regulation of vitamin D metabolism; the hypophosphatemia stimulates 25-hydroxyvitamin D-1α-hydroxylase activity and, as a consequence, 1,25-(OH)2D3 reaches a high serum level in our patients; (b) hypercalciuria with nephrocalcinosis and progressive renal impairment; the stimulation of intestinal calcium absorption by the 1,25-(OH)2D3 leads to an increase in the renal filtered calcium.
Hypercalciuria is also present in 7 unaffected members of our pedigree (fig. 1). A different inherited form of hypophosphatemic rickets with hypercalciuria (HHRH) was observed in 9 members of a Beduin tribe in which the phenotype segregates as an autosomal recessive trait [33, 34]. In that tribe, 21 asymptomatic members presented idiopathic hypercalciuria while the phosphorus serum level and excretion were intermediate between those found in patients and in normal members of the same tribe. The authors hypothesized that a single gene is responsible both for HHRH and hypercalciuria. The level of the phosphatemia appears to determine which subjects will have hypercalciuria alone and which will also have rickets.
In contrast, the 7 asymptomatic members of our pedigree show no other biochemical abnormality in addition to hypercalciuria. We therefore cannot postulate that a single gene is responsible in our pedigree for both XLRH and hypercalciuria. In this case, hypercalciuria is more likely to be caused by another gene which segregates independently of the new form of X-linked hypophosphatemic rickets. Hypercalciuria alone has been previously reported to segregate as an inherited trait in large pedigrees [35]. In order to test whether HPDR I and XLRH represent two different genes on the human X chromosome, we performed a linkage study in this pedigree using markers from the Xp22.1–p22.2 region where HPDR I is located. In this way, we excluded the whole region between DXS43 and DXS41 where HPDR I maps.
On the other hand, positive lod score values were obtained using traditional markers from the Xp11.22–p11.23 region. To increase the marker informativity within this candidate interval, we performed the same analysis using some recently identified highly polymorphic markers [25]. In this way, we mapped the XLRH gene to the Xp11.2 region at 0% recombination fraction from the DXS1039 locus.
We conclude therefore that XLRH in our pedigree and HPDR I are two genetically independent forms of X-linked hypophosphatemia. On the basis of sensorineural hearing deficits due to cochlear dysfunction and the X-linked dominant pattern of inheritance, the patients described by Boneh et al. [15] may represent the human counterpart of the Gyro phenotype in the mouse. However, these patients showed neither hypercalciuria nor a high level of 1,25-(OH)2D3 [C. Scriver, pers. commun.]. In contrast, in our pedigree, all the affected males showed hypercalciuria and high levels of 1,25-(OH)2D3, in addition to hypophosphatemia and rickets/osteomalacia. Inner ear involvement was documented in only one of these patients, while the other patients could not be examined. These clinical findings clearly indicate that the patients in our pedigree and those described by Boneh et al. [15] are affected with different disorders. This conclusion is in agreement with the apparent discrepancy between the X-linked dominant mode of transmission observed by Boneh et al. [15] and the X-linked recessive pattern of inheritance present in our pedigree.
It is interesting that X-linked recessive nephrolithiasis [36] was mapped to Xp11.22 using a large multigeneration pedigree [37]. This disease does not in any way affect the bone tissue, unlike the hypophosphatemias. A more recent mapping in Xp11.22 of a variant form of Fanconi syndrome, namely Dent disease [38], is even more interesting because some of the features of the latter disorder (hypophosphatemia, hypercalciuria, nephrocalcinosis and rickets) are found in the Italian pedigree studied in the present work. On the other hand some typical manifestations of Dent disease (generalized amminoaciduria, hypokalemia) suggesting that this is a generalized proximal tubular defect, are absent in the Italian pedigree which shows decreased phosphate-specific tubular reabsorption. Whether the phenotypes of Dent disease and of XLRH are caused by different genes or by different mutations in the same gene remains to be established.
Also remaining to be established is which of the hypophosphatemic phenotypes corresponds to the Gyro mouse. The definition of dominant inheritance in the Gyro mouse was based only on the observation of hypophosphatemia in carrier females presenting a milder phenotype than the affected males. Hypophosphatemia alone is also present in two carrier females in our pedigree, namely individuals No. 4 and 15 (fig. 1). It should be noted that the definition of the female phenotype at an X-linked locus can also be hampered by skewed lyonization.
On the basis of the few rearrangements which should have occurred in the human and mouse X chromosome during evolution, it is expected that the human equivalent of the Gyro mouse should map in the same region where HPDR I is localized [6]. Contrary to this expectation, the XLRH gene maps in a different position of the X chromosome. However, the hypothesis that only few inversions have separated the two chromosomes is based on the comparative mapping of a relatively small number of already identified conserved loci, whereas other rearrangements may have occurred during evolution [32].
In addition, only two clusters of loci on the long arm of the human X chromosome are kept in the same relative order in the middle portion of the mouse X chromosome. Since the mouse X chromosome does not have a short arm, the loci which are located on the short arm of the human X chromosome are necessarily rearranged in different positions on the mouse X.
In conclusion, only after a more detailed map of loci on the human and mouse X chromosomes is available, will it be possible to hypothesize additional intrachromosomal rearrangements which might indicate the most likely localization of the Gyro mouse equivalent on the human X chromosome.
References
Anast CS, Carpenter TO, Lyndon Key L: Metabolic bone disorders in children; in Avioli LV, Krane SM: Metabolic Bone Disease and Clinically Related Disorders. Philadelphia, Saunders, 1990.
Bell CL, Tenenhouse HS, Scriver CR: Primary cultures of renal epithelial cells from X-linked hypophosphatemic (Hyp) mice express defects in phosphate transport and vitamin D metabolism. Am J Hum Genet 1988;43:293–303
Dobre CV, Alvarez UM, Hruska KA: Primary culture of hypophosphatemic proximal tubule cells express defective adaptation to phosphate. J Bone Miner Res 1990;5:205.
Machler M, Freyd D, Gal A, Orth U, Wienker TF, Fanconi A, Shmid W: X-linked dominant hypophosphatemia is closely linked to DNA markers DXS41 and DXS43 at Xp22. Hum Genet 1986,73:271–275.
Read AP, Thakker RV, Mountford RC, Brenton DP, Davies M, Glorieux F, Harris R, Hendy GN, King A, McGlade S, Peacock CJ, Smith R, O’Riordan JLH: Mapping of human X-linked hypophosphatemic rickets by multilocus linkage analyses. Hum Genet 1986;73:267–270
Thakker RV, Read AP, Davies KE, Whyte MP, Weksberg R, Glorieux F, Davies M, Mountford RC, Harris R, King A, Kim GS, Fraser D, Kooh SW, O’Riordan JLH: Bridging markers defining the map position of X-linked hypophosphatemic rickets. J Med Genet 1987;24:756–760
Thakker RV, Davies KE, Read AP, Tippett P, Wooding C, Flint T, Wood S, Kruse TA, White MP, O’Riordan JLH: Linkage analysis of two cloned DNA sequences, DXS197 and DXS207, in hypophosphatemic rickets families. Genomics 1990;8:189–193
Econs MJ, Pericak-Vance MA, Betz H, Barlett RJ, Speer MC, Drezner MK: The human glycine receptor: A new probe that is linked to the X-linked hypophosphatemic rickets gene. Genomics 1990;7:439–441
Econs MJ, Barker DF, Speer MC, Pericak-Vance MA, Fain PR, Drezner MK: Multilocus mapping of the X-linked hypophosphatemic rickets gene. J Clin Endocrinol Metab 1992;75:201–206
Lyon MF, Scriver CR, Baker LRI, Tenenhouse HS, Kronick J, Mandla S: The Gy mutation: Another cause of X-linked hypophosphatemia in mouse. Proc Natl Acad Sci USA 1986;83:4899–4903
Silver LM, Nadeau JH, Klem J: Encyclopedia of the mouse genome II. Mamm Genome 1992;3:1–280
Eicher EM, Southard CR, Glorieux FH: Hypophosphatemia: Mouse model for human familial hypophosphatemic (vitamin D-resistant) rickets. Proc Natl Acad Sci USA 1976;73:4667–4671
Davidai GA, Nesbitt T, Drezner MK: Normal regulation of calcitriol production in Gy mice. J Clin Invest 1990;85:334–339
Tenenhouse HS, Meyer RA, Mandla S, Meyer MH, Gray RW: Renal phosphate transport and vitamin D metabolism in X-linked hypophosphatemic Gy mice: Response to phosphate deprivation. Endocrinology 1992;181:51–56
Boneh A, Reade TM, Scriver CR, Rishikof E: Audiometric evidence for two forms of X-linked hypophosphatemia in humans, apparent counterparts of Hyp and Gy mutations in mouse. Am J Med Genet 1987;27:997–1003
Enia G, Zoccali C, Bolino A, Romeo G: New X-linked hypophosphatemic rickets with hypercalciuria leading to progressive renal failure. Nephrol Dial Transplant 1992;7:757–758
Maniatis T, Fritsch EF, Sambrook J: Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, Cold Spring Harbor Laboratory Press, 1977.
Williamson R, Bowcock A, Kidd K, Pearson P, Schmidtke J, Ceverha P, Chipperfield M, Cooper DN, Coutelle C, Hewitt J, Klinger K, Langley K, Beckmann J, Tolley M, Maidak B: Report of the DNA committee and catgalogues of cloned and mapped genes, markers formatted for PCR and DNA polymorphisms. Cytogenet Cell Genet 1991;58:1624–1659
Petty EM, Carstens R, Bale AE: Ornithine transcarbamylase polymorphism detected by PCR introduction of Dral site. Nucleic Acids Res 1991;19:690.
Weber JL, Kwiter AE, May PE, Polymeropoulos MH, Ledbetter S: Dinucleotide repeat polymorphisms at the DXS453, DXS454, DXS458 loci. Nucleic Acids Res 1990;18:4037.
Vignal A, Gyapay G, Hazan J, Nguyen S, Dupraz C, Cheron N, Becuwe N, Tranchant M, Weissembach J: A non-radioactive multiplex procedure for genotyping of micro-satellites markers. Methods Mol Genet, in press.
Weber JL, May PE: Abundant class of human DNA polymorphisms can be typed using the polymerase chain reaction. Am J Hum Genet 1989;44:388–396
Edwards A, Civitello A, Hammond HA, Caskey CT: DNA typing and genetic mapping with trimeric and tetrameric tandem repeats. Am J Hum Genet 1991;49:746–756
Ott J: Analysis of Human Genetic Linkage. Baltimore, Johns Hopkins University Press, 1991.
Weissenbach J, Gyapay G, Dib C, Vignal A, Morissette J, Millasseau P, Vaysseix G, Lathrop M: A second generation linkage map of the human genome. Nature 1992;359:794–801
Rowe PS, Read AP, Mountford R, Benham F, Kruse T, Camerino G, Davies KE, O’Riordan JLH: Three DNA markers for hypophosphatemic rickets. Hum Genet 1992;89:539–542
Alitalo T, Kruse TA, Ahrens P, Albertsen HM, Eriksson AW, de la Chapelle A: Genetic mapping of 12 marker loci in the Xp22.3-p21.2 region. Hum Genet 1991;86:599–603
Ohno S: Sex Chromosomes and Sex-Linked Genes. Berlin, Springer, 1967.
O’Brien SJ, Womack JE, Lyons LA, Moore KJ, Jenkins NA, Copeland NG: Anchored reference loci for comparative genome mapping in mammals. Nature Genet 1993;3:103–111
Lyon MF: Evolution of the X chromosome. Nature 1990;348:585–586
Buckle VJ, Edwards JH, Evans GP, Jonasson JA, Lyon MF, Peters J, Searle AG: Comparative maps of human and mouse X chromosomes. Cytogenet Cell Genet 1985;40:594.
Davisson MT: X-linked genetic homologies between mouse and man. Genomics 1987;1:213–227
Tieder M, Modai D, Samuel R, Arie R, Halabe A, Bab I, Gabizon D, Liberman U: Hereditary hypophosphatemic rickets with hypercalciuria. N Engl J Med 1985;312:611–617
Tieder M, Modai D, Shaked U, Samuel R, Arie R, Halabe A, Maor J, Weissgarten J, Averbukh Z, Cohen N, Edelstein S, Liberman UA: Idiopathic hypercalciuria and hereditary hypophosphatemic rickets: Two phenotypical expressions of a common genetic defect. N Engl J Med 1987;316:125–129
Nordin BEC: Hypercalciuria. Clin Sci Mol Med 1977;52:1–8
Frymoyer PA, Scheinman SJ, Dunham PB, Jones DB, Hueber P, Schroeder ET: X-linked recessive nephrolithiasis with renal failure. N Engl J Med 1991;325:681–686
Scheinman SJ, Pook MA, Wooding C, Pang JT, Frymoyer PA, Thakker RV: Mapping the gene causing X-linked recessive nephrolithiasis to Xp11.22 by linkage studies. J Clin Invest 1993;91:2351–2357
Thakker RV, Pook MA, Wooding C, Norden AGW, Feest TG, Wrong OM: The gene causing Dent’s disease, a renal Fanconi syndrome with nephrocalcinosis and kidney stones, is on the short arm of the X chromosome (Xp11.22) (abstract 94). J Bone Miner Res 1993;8:140.
Acknowledgements
This work was supported by grants from the Italian CNR (Progetto Finalizzato Ingegneria Genetica) from the Italian Ministry of Health, from the Italian Telethon and from the Commission of the European Communities (CEC), proposal No. PL910027. The technical help of Mr. Francesco Caroli and Mr. Giuseppe Santamaria is gratefully acknowledged. The authors thank Dr. J. Ott for helpful discussion.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Bolino, A., Devoto, M., Enia, G. et al. Genetic Mapping in the Xp11.2 Region of a New Form of X-Linked Hypophosphatemic Rickets. Eur J Hum Genet 1, 269–279 (1993). https://doi.org/10.1159/000472424
Received:
Revised:
Accepted:
Issue Date:
DOI: https://doi.org/10.1159/000472424
Key Words
This article is cited by
-
Genetics and phenotypic heterogeneity of Dent disease: the dark side of the moon
Human Genetics (2021)
-
Phenotype of dent disease in a cohort of Indian children
Indian Pediatrics (2016)
-
Dent’s disease: clinical features and molecular basis
Pediatric Nephrology (2011)
-
Vitamin A responsive night blindness in Dent’s disease
Pediatric Nephrology (2009)
-
Mechanisms of Disease: what can mouse models tell us about the molecular processes underlying Dent disease?
Nature Clinical Practice Nephrology (2007)
