
Abstract
In contrast with the reported almost exclusive paternal origin of de novo mutations in MEN 2A, FMTC and MEN 2B, de novo mutations in Hirschsprung patients arise both on paternal and maternal chromosomes. This distinctive feature of RET mutations associated with Hirschsprung’s disease and of the RET mutations associated with thyroid cancer indicates a basic biological difference between the mutational events leading to the different phenotypes.
Similar content being viewed by others

Hirschsprung disease (HSCR) is a frequent congenital disorder (with an incidence of approximately 1 in every 5,000 live births) appearing during the first years of life and characterized by the absence of parasympathetic intrinsic ganglion cells in the hindgut. A major gene for HSCR with incompletely penetrant autosomal dominant mode of inheritance was mapped to the proximal long arm of chromosome 10 [1, 2] and subsequently restricted by genetic and physical mapping to the RET locus [3], in which point mutations as well as deletion and insertion mutations were identified [4, 5]. Among these mutations, a proportion varying between 21 and 26% [6–9] can be estimated to be de novo, which is in keeping with the high incidence of sporadic cases (80–90%) observed among HSCR patients.
HSCR mutations are distributed among 18 of the 21 exons of RET [9]. On the contrary, mutations causing multiple endocrine neoplasia type 2A (MEN 2A) are exclusively found in exons 10 and 11 [10, 11], those causing familial medullary thyroid carcinoma (FMTC) mostly in exons 10, 11 and rarely in exons 13 and 14 [12, 13], while a unique mutation in codon 918 (Met→Thr) of exon 16 is found in almost all multiple endocrine neoplasia type 2B (MEN 2B) patients [14, 15]. The latter one frequently arises as a de novo event almost exclusively on the paternal chromosome [16, 17], with skewed segregation in subsequent generations, a fact arguing in favor of genomic imprinting [18]. In addition a recent study indicates that up to 9% of MEN 2A and FMTC patients have de novo mutations which occur exclusively on the paternal allele [19]. In our series of 121 HSCR patients, we identified 23 RET mutations, 6 in familial and 17 in sporadic cases [6, 9]. Of the latter ones, in 6 cases a parent carried the mutation, in 6 the mutation occurred de novo, while investigations of the other 5 were inconclusive because of lack of parental DNA. Thus between 35% (6/17) and 65% (11/17) of RET mutations in sporadic HSCR patients occur de novo, which represents a prevalence higher than that previously calculated [6–9].
Table 1 summarizes investigations of the 6 de novo mutations. In patients 1 and 2 the RET exon containing the mutation [6] also carried an informative polymorphism [20], allowing the parental origin to be established. Parental origin in patients 3 and 4 [6, 9] was established by isolating the normal and abnormal chromosomes in somatic cell hybrids [21] and typing for informative polymorphisms [22–24] both parents and the affected child, together with hybrid clones (fig. 1). Patient 5 [25] had a de novo deletion of the entire RET gene, with loss of paternal allele at the linked marker D10S196. No conclusion could be reached in patient 6 because of lack of informative polymorphisms.

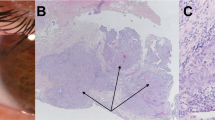
Parental origin analysis for the del3118(CTGG) de novo mutation. Normal and mutated RET homologues on chromosome 10 were segregated into distinct somatic cell hybrids after fusion of the patient’s lymphoblasts with a Chinese hamster ovary cell line (YH21). Hybrid cells containing chromosome 10 were selected for the expression of the gene coding for the beta subunit of the fibronectin receptor (FNRB), which maps in 10p11.2, using a monoclonal antibody against FNRB [21]. Hybrids 1 and 2 retain the normal and the deleted alleles of RET, respectively. A Pattern (on a 6% Polyacrylamide gel) of the PCR product of exon 19 of RET amplified from genomic DNA of the patient and his parents, as well as from 2 hybrids. The lower band represents the deleted allele, while the upper band represents the normal allele. B Typing obtained with the RET-INT5 microsatellite [23]. The genotypes of the father, of the affected son and of the mother are 4/6, 4/5 and 5/5, respectively. Hybrid 1 retains the normal RET and allele 5 of RET-INT5 derived from the mother, while hybrid 2 retains the deleted RET and allele 4 of RET-INT5 derived from the father.
In conclusion, de novo mutations of RET occur in both maternal (3/5) and paternal (2/5) alleles of HSCR patients, in contrast with MEN 2B [16, 17], MEN 2A and FMTC [19], in which de novo RET mutations arise almost exclusively on paternal chromosomes. It remains to be established on a larger series of de novo HSCR mutations (a) whether the observed breakdown (point mutations of maternal and deletions of paternal origin) is due to chance or has any biological meaning, and (b) how often the somatic mosaicism already reported in 1 HSCR patient [8] is present in patients carrying de novo mutations.
References
Lyonnet S, Bolino A, Pelet A, Abel L, Nihoul-Fékété C, Briard ML, Mok-Siu V, Kääriäinen H, Martucciello G, Lerone M, Puliti A, Yin L, Weissenbach J, Devoto M, Romeo G: A gene for Hirschsprung disease maps to the proximal long arm of chromosome 10. Nature Genet 1993;4:346–350
Angrist M, Kauffman E, Slaugenhaupt SA, Malise TC, Puffenberger EG, Washington SS, Lipson A, Cass DT, Reyna T, Weeks DE, Sieber W, Chakravarti A: A gene for Hirschsprung disease (megacolon) in the pericentromeric region of human chromosome 10. Nature Genet 1993;4:351–356
Yin L, Ceccherini I, Pasini B, Matera I, Bicocchi MP, Barone V, Bocciardi R, Kääriäinen H, Weber D, Devoto M, Romeo G: Close linkage with the RET proto-oncogene and boundaries of deletion mutations in autosomal dominant Hirschsprung disease. Hum Mol Genet 1993;2:1803–1808
Romeo G, Ronchetto P, Yin L, Barone V, Seri M, Ceccherini I, Pasini B, Bocciardi R, Lerone M, Kääriäinen H, Martucciello G: Point mutations affecting the tyrosine kinase domain of the RET proto-oncogene in Hirschsprung’s disease. Nature 1994;367:377–378
Edery P, Lyonnet S, Mulligan LM, Pelet A, Dow E, Abel L, Holder S, Nihoul-Fékété C, Ponder BAJ, Munnich A: Mutations of the RET proto-oncogene in Hirschsprung’s disease. Nature 1994;367:378–380
Yin L, Barone V, Seri M, Bolino A, Bocciardi R, Ceccerini I, Pasini B, Tocco T, Lerone M, Cywes S, Moore S, Vanderwinden JM, Abramowics MJ, Kristoffersson U, Larsson LT, Hamel BCJ, Silengo M, Martucciello G, Romeo G: Heterogeneity and low detection rate of RET mutations in Hirschsprung disease. Eur J Hum Genet 1994;2:272–280
Angrist M, Bolk S, Thiel B, Puffenberger EG, Hofstra RM, Buys CHCM, Cass DT, Chakravarti A: Mutations analysis of the RET receptor tyrosine kinase in Hirschsprung disease. Hum Mol Genet 1995;4:821–830
Attié T, Pelet A, Edery P, Eng C, Mulligan LM, Amiel J, Boutrand L, Beldjord C, Nihoul-Fékété C, Munnich A, Ponder BAJ, Lyonnet S: Diversity of RET proto-oncogene mutations in familial and sporadic Hirschsprung disease. Hum Mol Genet 1995;4:1381–1386
Seri M, Yin L, Barone V, Bolino A, Celli I, Bocciardi R, Pasini B, Ceccherini I, Lerone M, Kristoffersson U, Larsson LT, Casasa JM, Cass DT, Abramowicz MJ, Vanderwinden JM, Kravcenkiene I, Baric I, Silengo M, Martucciello G, Romeo G: Detection of RET mutations is higher among long segment than short segment Hirschsprung patients. Hum Mutat, in press.
Mulligan LM, Kwok JBJ, Healey CS, Elsdon MJ, Eng C, Gardner E, Love DR, Mole SE, Moore JK, Papi L, Ponder MA, Telenius H, Tunnacliffe A, Ponder BAJ: Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature 1993;363:458–460
Donis-Keller H, Dou S, Chi D, Carlson KM, Toshima K, Lairmore TC, Howe JR, Moley JF, Goodfellow P, Wells A: Mutations in the RET proto-oncogene are associated with MEN 2A and FMTC. Hum Mol Genet 1993;2:851–856
Eng C, Smith DP, Mulligan LM, Healey CS, Zvelebil MJ, Stonehouse TJ, Ponder MA, Jackson CE, Waterfield MD, Ponder BAJ: A novel point mutation in the tyrosine kinase domain of the RET proto-oncogene in sporadic medullary thyroid carcinoma and in a family with FMTC. Oncogene 1995;10:509–513
Bolino A, Schuffenecker I, Yin L, Seri M, Silengo M, Tocco T, Chabrier G, Houdent C, Murat A, Schlumberger M, Tourniaire J, Lenoir GM, Romeo G: RET mutations in exons 13 and 14 of FMTC patients. Oncogene 1995;10:2415–2419
Hofstra RMW, Landsvater RM, Ceccherini I, Stulp RP, Stelwagen T, Yin L, Pasini B, Höppener JWM, Van Amstel HKP, Romeo G, Lips CJM, Buys CHCM: A mutation in the RET proto-oncogene associated with multiple endocrine neoplasia type 2B and sporadic medullary thyroid carcinoma. Nature 1994;367:375–376
Carlson KM, Dou S, Chi D, Scavarda N, Toshima K, Jackson CE, Wells SA, Goodfellow PJ, Donis-Keller H: Single missense mutation in the tyrosine kinase catalytic domain of the RET protooncogene is associated with multiple endocrine neoplasia type 2B. Proc Natl Acad Sci USA 1994;91:1579–1583
Carlson KM, Bracamontes J, Jackson CE, Clark R, Lacroix A, Wells SA, Goodfellow PJ: Parent-of-origin effects in multiple endocrine neoplasia type 2B. Am J Hum Genet 1994;55:1076–1082
Kitamura Y, Scavarda N, Wells SA, Jackson CE, Goodfellow PJ: Two maternally derived missense mutations in the tyrosine kinase domain of the RET protooncogene in a patient with de novo MEN 2B. Hum Mol Genet 1995;4:1987–1988
Sapienza C: Parental origin effects, genome imprinting, and sex-ratio distortion: Double or nothing? Am J Hum Genet 1994;55:1073–1075
Schuffenecker I, Ginet N, Goldgar D, Eng C, Chambe B, Boneu A, Houdent C, Pallo D, Schlumberger M, Thivolet C, Lenoir GM: Prevalence and parental origin of de novo RET mutations in MEN 2A and FMTC. Am J Hum Genet, in press.
Ceccherini I, Hofstra RMW, Yin L, Stulp RP, Barone V, Stelwagen T, Bocciardi R, Nijveen H, Bolino A, Seri M, Ronchetto P, Pasini B, Bozzano M, Buys CHCM, Romeo G: DNA polymorphisms and conditions for SSCP analysis of the 20 exons of the RET proto-oncogene. Oncogene 1994;9:3025–3030
Puliti A, Covone AE, Bicocchi MP, Bolino M, Martucciello G, Jasonni V, Romeo G: Deleted and normal chromosome 10 homologs from a patient with Hirschsprung disease isolated in two cell hybrids through enrichment by immunomagnetic selection. Cytogenet Cell Genet 1993;63:102–106
Gardner E, Papi L, Easton DF, Cummings T, Jackson CE, Kaplan M, Love DR, Mole SE, Moore JK, Mulligan LM, Norum RA, Ponder MA, Reichlin S, Stall G, Telenius H, Telenius-Berg M, Tunnacliffe A, Ponder BAJ: Genetic linkage studies map the multiple endocrine neoplasia type 2 loci to a small interval on chromosome 10q11.2. Hum Mol Genet 1993;2:241–246
Pasini B, Hofstra RMW, Yin L, Bocciardi R, Santamaria G, Grootscholten PM, Ceccherini I, Patrone G, Priolo M, Buys CHCM, Romeo G: The physical map of the human RET protooncogene. Oncogene 1995;11:1737–1743
Lairmore TC, Dou S, Howe JR, Chi D, Carlson K, Velle R, Mishra SK, Wells SA, Donis-Keller H: A 1.5-megabase yeast artificial chromosome contig from human chromosome 10q11.2 connecting three genetic loci (RET, D10S94, and D10S102) closely linked to the MEN 2A locus. Proc Natl Acad Sci USA 1989;90:492–496
Martucciello G, Bicocchi MP, Dodero P, Lerone M, Cirillo MS, Puliti A, Gimelli G, Romeo G, Jasonni V: Total colonic aganglionosis associated with interstitial deletion of the long arm of chromosome 10. Pediatr Surg Int 1992;7:308–310
Acknowledgements
This work was supported by grants from the Italian Ministry of Health, the Italian CNR (Progetto Finalizzato Ingeneria Genetica). Y.L. was a recipient of a fellowship from Italian TELETHON.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Yin, L., Seri, M., Barone, V. et al. Prevalence and Parental Origin of de novo RET Mutations in Hirschsprung’s Disease. Eur J Hum Genet 4, 356–358 (1996). https://doi.org/10.1159/000472232
Received:
Revised:
Accepted:
Issue Date:
DOI: https://doi.org/10.1159/000472232
Key Words
This article is cited by
-
Novel MLPA procedure using self-designed probes allows comprehensive analysis for CNVs of the genes involved in Hirschsprung disease
BMC Medical Genetics (2010)
-
A novel study of Copy Number Variations in Hirschsprung disease using the Multiple Ligation-dependent Probe Amplification (MLPA) technique
BMC Medical Genetics (2009)
-
Genetic basis of Hirschsprung’s disease
Pediatric Surgery International (2009)
-
Molekularbiologie, Grundlagenforschung und Diagnose des Morbus Hirschsprung
Der Pathologe (2007)
