
Featured
-
-
Article
| Open Access High-throughput mechanical phenotyping and transcriptomics of single cells
High-throughput mechanical phenotyping and transcriptomics of single cells
The molecular system regulating cell surface mechanics remains largely unexplored at single-cell resolution. Here, the authors report a high-throughput single-cell assay, ELASTomics, which integrates mechanical phenotyping with unbiased transcriptomics.
- Akifumi Shiomi
- , Taikopaul Kaneko
- & Hirofumi Shintaku
-
Article
| Open Access Enzyme-assisted high throughput sequencing of an expanded genetic alphabet at single base resolution
Enzyme-assisted high throughput sequencing of an expanded genetic alphabet at single base resolution
The expansion of the genetic code with synthetic nucleotides has broadened our ability to evolve DNA as a functional material, but we lack analytical tools for the expanded alphabet. Here the authors demonstrate an enzyme-assisted method for the sequencing of six-letter DNA.
- Bang Wang
- , Kevin M. Bradley
- & Steven A. Benner
-
Article
| Open Access GRouNdGAN: GRN-guided simulation of single-cell RNA-seq data using causal generative adversarial networks
GRouNdGAN: GRN-guided simulation of single-cell RNA-seq data using causal generative adversarial networks
Benchmarking GRN inference methods remains a challenge. Here, authors present GRouNdGAN, a causal generative model that imposes a user-defined GRN in its architecture to simulate realistic single-cell data, bridging the gap between synthetic and biological data benchmarks of GRN inference methods.
- Yazdan Zinati
- , Abdulrahman Takiddeen
- & Amin Emad
-
Article
| Open Access Transfer learning enables identification of multiple types of RNA modifications using nanopore direct RNA sequencing
Transfer learning enables identification of multiple types of RNA modifications using nanopore direct RNA sequencing
Simultaneous profiling of multiple RNA modifications is a promising yet understudied field of research. Here, authors develop a transferable deep learning framework capable of detecting multiple types of RNA modifications in single nanopore sequencing sample.
- You Wu
- , Wenna Shao
- & Xiang Yu
-
Article
| Open Access Multiplexed bulk and single-cell RNA-seq hybrid enables cost-efficient disease modeling with chimeric organoids
Multiplexed bulk and single-cell RNA-seq hybrid enables cost-efficient disease modeling with chimeric organoids
IPSC-derived organoids model diseases. Multiplexed coculture and demultiplexing natural genetic barcodes aid in studying genetic effects. Here, authors introduce Vireo-bulk to deconvolve bulk RNA-seq data, quantify donor abundance and identify differentially expressed genes.
- Chen Cheng
- , Gang Wang
- & Jin Zhang
-
Article
| Open Access Prediction of m6A and m5C at single-molecule resolution reveals a transcriptome-wide co-occurrence of RNA modifications
Prediction of m6A and m5C at single-molecule resolution reveals a transcriptome-wide co-occurrence of RNA modifications
The epitranscriptome holds many unexplored RNA functions, but detecting multiple modifications from one sample remains challenging. Here, authors devise a strategy combining AI and nanopore sequencing to uncover a transcriptome-wide co-occurrence of two modification types in individual RNA molecules.
- P Acera Mateos
- , A J Sethi
- & E Eyras
-
Article
| Open Access scLENS: data-driven signal detection for unbiased scRNA-seq data analysis
scLENS: data-driven signal detection for unbiased scRNA-seq data analysis
Single-cell RNA sequencing data analysis is limited by noise and high dimensionality. Here, authors present scLENS, a tool that automates accurate signal detection without manual input, particularly in complex datasets.
- Hyun Kim
- , Won Chang
- & Jae Kyoung Kim
-
Article
| Open Access High clonal diversity and spatial genetic admixture in early prostate cancer and surrounding normal tissue
High clonal diversity and spatial genetic admixture in early prostate cancer and surrounding normal tissue
It remains challenging to characterise somatic copy number alterations (SCNAs) in tumors and the surrounding tissues with spatial and single-cell resolution. Here, the authors develop the scCUTseq approach to characterise SCNAs from single cells in multi-region prostate cancer samples and identify pseudo-diploid cells and subclones.
- Ning Zhang
- , Luuk Harbers
- & Nicola Crosetto
-
Article
| Open Access scButterfly: a versatile single-cell cross-modality translation method via dual-aligned variational autoencoders
scButterfly: a versatile single-cell cross-modality translation method via dual-aligned variational autoencoders
Technical limitations of simultaneously multi-omics profiling lead to highly noisy multi-modal data and substantial costs. Here, authors proposed a versatile framework and data augmentation schemes, capable of single-cell cross-modality translation and multiple extensive applications.
- Yichuan Cao
- , Xiamiao Zhao
- & Shengquan Chen
-
Article
| Open Access Pianno: a probabilistic framework automating semantic annotation for spatial transcriptomics
Pianno: a probabilistic framework automating semantic annotation for spatial transcriptomics
Recognising spatial spots’ biological identity in spatial transcriptomics remains a challenge. Here, authors introduce Pianno, a tool that helps annotate the biological structures or cell-type constructions across diverse tissues, offering new perspectives on understanding spatial transcriptomics.
- Yuqiu Zhou
- , Wei He
- & Ying Zhu
-
Article
| Open Access FinaleMe: Predicting DNA methylation by the fragmentation patterns of plasma cell-free DNA
FinaleMe: Predicting DNA methylation by the fragmentation patterns of plasma cell-free DNA
DNA methylation from cell-free DNA (cfDNA) can be profiled using whole genome bisulfite sequencing (WGBS). Here, the authors develop a computational method, FinaleMe, that predicts DNA methylation and tissues of-origin in cfDNA and validate its performance using paired deep and shallow-coverage whole-genome sequencing (WGS) and WGBS data.
- Yaping Liu
- , Sarah C. Reed
- & Manolis Kellis
-
Article
| Open Access PLMSearch: Protein language model powers accurate and fast sequence search for remote homology
PLMSearch: Protein language model powers accurate and fast sequence search for remote homology
Homologous protein search is one of the most commonly used methods for protein analysis. Here, authors propose PLMSearch, a search method that takes only sequences as input and can search millions of protein pairs in seconds while maintaining sensitivity comparable to SOTA structure search methods.
- Wei Liu
- , Ziye Wang
- & Shanfeng Zhu
-
Article
| Open Access DELVE: feature selection for preserving biological trajectories in single-cell data
DELVE: feature selection for preserving biological trajectories in single-cell data
Characteristic genes or proteins driving continuous biological processes are difficult to uncover from noisy single-cell data. Here, authors present DELVE, an unsupervised feature selection method to identify core molecular features driving cell fate decisions.
- Jolene S. Ranek
- , Wayne Stallaert
- & Jeremy E. Purvis
-
Article
| Open Access A universal molecular control for DNA, mRNA and protein expression
A universal molecular control for DNA, mRNA and protein expression
Multi-omics analyses powerfully combine gene expression and translation, however no available controls can be used across these techniques. Here the authors develop pREF, a universal control construct designed for use in DNA, RNA and protein analyses.
- Helen M. Gunter
- , Scott E. Youlten
- & Tim R. Mercer
-
Article
| Open Access NAP-seq reveals multiple classes of structured noncoding RNAs with regulatory functions
NAP-seq reveals multiple classes of structured noncoding RNAs with regulatory functions
The genome-wide prevalence, mechanism and function of noncapped RNAs (napRNAs) are currently poorly understood. Here, the authors develop a method called NAP-seq, to globally profile the full-length sequences of napRNAs, revealing several classes of structured noncoding RNAs.
- Shurong Liu
- , Junhong Huang
- & Jianhua Yang
-
Article
| Open Access Targeted metagenomics reveals association between severity and pathogen co-detection in infants with respiratory syncytial virus
Targeted metagenomics reveals association between severity and pathogen co-detection in infants with respiratory syncytial virus
The impact of other pathogens on disease outcome was studied in European infants with RSV infection. Additional viruses were commonly co-detected during infection but were weakly linked to severity. However, presence of Haemophilus bacteria strongly associated with severe cases.
- Gu-Lung Lin
- , Simon B. Drysdale
- & Andrew J. Pollard
-
Article
| Open Access Single-cell division tracing and transcriptomics reveal cell types and differentiation paths in the regenerating lung
Single-cell division tracing and transcriptomics reveal cell types and differentiation paths in the regenerating lung
This study uses single-cell transcriptomics to examine how lung cells respond to targeted damage. The authors employ genetically modified mouse models and cell sorting to enrich for rare, actively dividing cells, revealing cell types/states and alternative differentiation paths.
- Leila R. Martins
- , Lina Sieverling
- & Claudia Scholl
-
Article
| Open Access An organism-wide atlas of hormonal signaling based on the mouse lemur single-cell transcriptome
An organism-wide atlas of hormonal signaling based on the mouse lemur single-cell transcriptome
Endocrinologists have traditionally focused on studying one hormone or organ system at a time. Here the authors use transcriptomic data from the mouse lemur to globally characterize primate hormonal signaling, describing hormone sources and targets, identifying conserved and primate specific regulation, and elucidating principles of the network.
- Shixuan Liu
- , Camille Ezran
- & James E. Ferrell Jr.
-
Article
| Open Access ProTInSeq: transposon insertion tracking by ultra-deep DNA sequencing to identify translated large and small ORFs
ProTInSeq: transposon insertion tracking by ultra-deep DNA sequencing to identify translated large and small ORFs
Identifying small proteins is challenging. ProTInSeq uses modified transposons to express markers inserted in-frame to protein-coding genes. This method identifies 153 unannotated small proteins in M. pneumoniae and additional proteomic information.
- Samuel Miravet-Verde
- , Rocco Mazzolini
- & Luis Serrano
-
Article
| Open Access Gene expression analyses reveal differences in children’s response to malaria according to their age
Gene expression analyses reveal differences in children’s response to malaria according to their age
Here the authors use dual RNA sequencing to characterize host and parasite gene expression from 136 Malian children with symptomatic Plasmodium falciparum infection. They find that parasitemia levels correlate with neutrophil and T cell levels and that the child’s age correlates with innate immune gene expression as well as gametocyte levels.
- Kieran Tebben
- , Salif Yirampo
- & David Serre
-
Article
| Open Access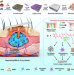 Endogenous stimuli-responsive separating microneedles to inhibit hypertrophic scar through remodeling the pathological microenvironment
Endogenous stimuli-responsive separating microneedles to inhibit hypertrophic scar through remodeling the pathological microenvironment
The treatment of hypertrophic scar (HS) is hindered by the low bioavailability of drugs and the pathological microenvironment. Here the authors report a separating microneedle drug delivery system responsive to high reactive oxygen species levels and overexpression of matrix metalloproteinases to remodel the pathological microenvironment for HS treatment.
- Zhuo-Ran Yang
- , Huinan Suo
- & Jintao Zhu
-
Article
| Open Access A single-cell atlas of Drosophila trachea reveals glycosylation-mediated Notch signaling in cell fate specification
A single-cell atlas of Drosophila trachea reveals glycosylation-mediated Notch signaling in cell fate specification
Studying Drosophila trachea development can inform the mechanisms of growth of all tubular structures. Here, the authors generate a transcriptomic cell atlas of the developing fly trachea and establish roles for Notch signaling, which may be disrupted by diet-induced glycosylation.
- Yue Li
- , Tianfeng Lu
- & Hai Huang
-
Article
| Open Access Riboformer: a deep learning framework for predicting context-dependent translation dynamics
Riboformer: a deep learning framework for predicting context-dependent translation dynamics
Riboformer is a deep learning-based framework that predicts changes in translation dynamics with codon-level precision. It corrects experimental artifacts in ribosome profiling data and identifies sequences causing ribosome stalling.
- Bin Shao
- , Jiawei Yan
- & Allen R. Buskirk
-
Article
| Open Access scCircle-seq unveils the diversity and complexity of extrachromosomal circular DNAs in single cells
scCircle-seq unveils the diversity and complexity of extrachromosomal circular DNAs in single cells
Extrachromosomal circular DNAs (eccDNAs) affect gene expression and tumour progression. Here, the authors report a method, scCircle-seq, for eccDNA profiling in single cells, demonstrating the stochasticity, cell type specificity, and dynamics of eccDNAs in cell lines and primary tumour samples.
- Jinxin Phaedo Chen
- , Constantin Diekmann
- & Nicola Crosetto
-
Article
| Open Access scCASE: accurate and interpretable enhancement for single-cell chromatin accessibility sequencing data
scCASE: accurate and interpretable enhancement for single-cell chromatin accessibility sequencing data
Single-cell chromatin accessibility sequencing (scCAS) data suffers from high sparsity and dimensionality. Here, authors propose an accurate and interpretable computational framework for enhancing scCAS data that considers cell-to-cell similarity.
- Songming Tang
- , Xuejian Cui
- & Shengquan Chen
-
Article
| Open Access TFvelo: gene regulation inspired RNA velocity estimation
TFvelo: gene regulation inspired RNA velocity estimation
Most RNA velocity models extract dynamics from the phase delay between unspliced and spliced mRNA for each gene. Here, authors propose TFvelo, broadening RNA velocity beyond splicing information to include gene regulation. TFvelo accurately models genes dynamics and infers cell pseudo-time from RNA abundance data.
- Jiachen Li
- , Xiaoyong Pan
- & Hong-Bin Shen
-
Article
| Open Access Predicting proximal tubule failed repair drivers through regularized regression analysis of single cell multiomic sequencing
Predicting proximal tubule failed repair drivers through regularized regression analysis of single cell multiomic sequencing
A profibrotic, proinflammatory kidney cell population has been identified as a driver of chronic kidney disease. Here, authors generate a human kidney single cell multiomic dataset and apply a regularised regression approach to identify transcription factors underpinning this cell population.
- Nicolas Ledru
- , Parker C. Wilson
- & Benjamin D. Humphreys
-
Article
| Open Access The dynamic genetic determinants of increased transcriptional divergence in spermatids
The dynamic genetic determinants of increased transcriptional divergence in spermatids
Here the authors show that genetic changes between species often alter gene expression in a cell type-specific manner. Most of this variability is driven by locally functioning cis-acting variation, and this contributes to the speed at which cell types accumulate expression changes.
- Jasper Panten
- , Tobias Heinen
- & Duncan T. Odom
-
Article
| Open Access Eco-evolutionary dynamics of gut phageome in wild gibbons (Hoolock tianxing) with seasonal diet variations
Eco-evolutionary dynamics of gut phageome in wild gibbons (Hoolock tianxing) with seasonal diet variations
The significance of gut phageome for wild animals with seasonal diets remains unexplored. Here, the authors use complementary metagenomics to analyze the phage-host dynamics and its implications for diet variations in wild skywalker hoolock gibbons.
- Shao-Ming Gao
- , Han-Lan Fei
- & Peng-Fei Fan
-
Article
| Open Access Mechanistic characterization of a Drosophila model of paraneoplastic nephrotic syndrome
Mechanistic characterization of a Drosophila model of paraneoplastic nephrotic syndrome
The fruit fly Drosophila melanogaster has emerged as a model to characterize the mechanisms of tumor-induced host organ dysfunction. Here, Xu, Liu et al. describe a mechanism of tumor-induced kidney dysfunction through hyper-activation of the PvR/JNK/Jra pathway in the Principal cells of the fly kidney/Malpighian tubules.
- Jun Xu
- , Ying Liu
- & Norbert Perrimon
-
Article
| Open Access A method to estimate the contribution of rare coding variants to complex trait heritability
A method to estimate the contribution of rare coding variants to complex trait heritability
The contribution of rare variants to complex traits has not been well studied. Here, the authors present RARity, a method to assess rare variant heritability without assuming a particular genetic architecture and enabling both gene-level and exome-wide heritability estimation of continuous traits.
- Nazia Pathan
- , Wei Q. Deng
- & Guillaume Paré
-
Article
| Open Access Semi-supervised integration of single-cell transcriptomics data
Semi-supervised integration of single-cell transcriptomics data
Batch effects hinder multi-sample single-cell data analyses. Here, authors present STACAS, a scalable single-cell RNA-seq data integration tool that uses prior cell type knowledge to preserve biological variability, demonstrating robustness to noisy input cell type labels.
- Massimo Andreatta
- , Léonard Hérault
- & Santiago J. Carmona
-
Article
| Open Access Utility of long-read sequencing for All of Us
Utility of long-read sequencing for All of Us
Using All of Us pilot data, the authors compared short- and long-read performance across medically relevant genes and showcased the utility of long reads to improve variant detection and phasing in easy and hard to resolve medically relevant genes.
- M. Mahmoud
- , Y. Huang
- & F. J. Sedlazeck
-
Article
| Open Access Human whole-exome genotype data for Alzheimer’s disease
Human whole-exome genotype data for Alzheimer’s disease
The heterogeneity of whole-exome sequencing (WES) data generation methods presents a challenge to joint analysis. Here, the authors present a bioinformatics strategy to generate high-quality data from processing diversely generated WES samples, as applied in the Alzheimer’s Disease Sequencing Project.
- Yuk Yee Leung
- , Adam C. Naj
- & Li-San Wang
-
Article
| Open Access PROST: quantitative identification of spatially variable genes and domain detection in spatial transcriptomics
PROST: quantitative identification of spatially variable genes and domain detection in spatial transcriptomics
Understanding biological mechanisms requires a thorough exploration of spatiotemporal transcriptional patterns in complex tissues. Here, authors present PROST to quantify spatial gene expression patterns and detect spatial domains using spatial transcriptomics data of varying resolutions.
- Yuchen Liang
- , Guowei Shi
- & Zhonghui Tang
-
Article
| Open Access Leveraging single-cell ATAC-seq and RNA-seq to identify disease-critical fetal and adult brain cell types
Leveraging single-cell ATAC-seq and RNA-seq to identify disease-critical fetal and adult brain cell types
This study analyzed data from human cells assayed using single-cell technologies, together with data associating genetic variants to disease, to identify fetal and brain cell types whose biologically critically influences the etiology of disease.
- Samuel S. Kim
- , Buu Truong
- & Alkes L. Price
-
Article
| Open Access BIDCell: Biologically-informed self-supervised learning for segmentation of subcellular spatial transcriptomics data
BIDCell: Biologically-informed self-supervised learning for segmentation of subcellular spatial transcriptomics data
Subcellular in situ spatial transcriptomics offers the promise to address biological problems that were previously inaccessible but requires accurate cell segmentation to uncover insights. Here, authors present BIDCell, a biologically informed, deep learning-based cell segmentation framework.
- Xiaohang Fu
- , Yingxin Lin
- & Jean Y. H. Yang
-
Article
| Open Access Direct RNA sequencing coupled with adaptive sampling enriches RNAs of interest in the transcriptome
Direct RNA sequencing coupled with adaptive sampling enriches RNAs of interest in the transcriptome
It can be difficult to find rare transcripts when sequencing a transcriptome. Here the authors show adaptive sampling on direct RNA runs to increase the likelihood of finding less frequent ones while selectively ejecting the higher-abundance transcripts.
- Jiaxu Wang
- , Lin Yang
- & Yue Wan
-
Article
| Open Access VESPA: an optimized protocol for accurate metabarcoding-based characterization of vertebrate eukaryotic endosymbiont and parasite assemblages
VESPA: an optimized protocol for accurate metabarcoding-based characterization of vertebrate eukaryotic endosymbiont and parasite assemblages
DNA sequencing methods for characterizing microbial communities are well developed for bacteria, archaea and fungi, but less so for eukaryotic parasites and commensals. Here, the authors present an optimized and validated metabarcoding protocol for host-associated eukaryotic communities.
- Leah A. Owens
- , Sagan Friant
- & Tony L. Goldberg
-
Article
| Open Access MarsGT: Multi-omics analysis for rare population inference using single-cell graph transformer
MarsGT: Multi-omics analysis for rare population inference using single-cell graph transformer
Identifying rare cell populations is key to understanding cancer progression and response to therapy. Here, authors introduce MarsGT, an end-to-end deep learning model for rare cell population identification from scMulti-omics data.
- Xiaoying Wang
- , Maoteng Duan
- & Qin Ma
-
Article
| Open Access MENDER: fast and scalable tissue structure identification in spatial omics data
MENDER: fast and scalable tissue structure identification in spatial omics data
Identifying tissue structure in large-scale spatial omics datasets from multiple slices is challenging. Here, authors present MENDER, an optimisation-free spatial clustering method that can scale to million-level spatial data, enabling efficient analysis of spatial cell atlases.
- Zhiyuan Yuan
-
Article
| Open Access Angiogenesis-on-a-chip coupled with single-cell RNA sequencing reveals spatially differential activations of autophagy along angiogenic sprouts
Angiogenesis-on-a-chip coupled with single-cell RNA sequencing reveals spatially differential activations of autophagy along angiogenic sprouts
The functional heterogeneity of autophagy in endothelial cells during angiogenesis remains incompletely understood. Here, the authors apply a 3D angiogenesis-on-a-chip coupled with single-cell RNA sequencing to find distinct autophagy functions in two different endothelial cell populations during angiogenic sprouting.
- Somin Lee
- , Hyunkyung Kim
- & Noo Li Jeon
-
Article
| Open Access Terminal modifications independent cell-free RNA sequencing enables sensitive early cancer detection and classification
Terminal modifications independent cell-free RNA sequencing enables sensitive early cancer detection and classification
Cell free RNA is a potentially valuable resource to detect cancer, however, its low concentration in plasma can limit usefulness. Here, the authors devise a library preparation method from 100ul of plasma, and apply to multiple cancer types to detect and classify cancer patients
- Jun Wang
- , Jinyong Huang
- & Deming Gou
-
Article
| Open Access ECOLE: Learning to call copy number variants on whole exome sequencing data
ECOLE: Learning to call copy number variants on whole exome sequencing data
Copy number variants (CNV) are shown to contribute to the etiology of various genetic disorders. Here, authors present ECOLE, a deep learning-based somatic and germline CNV caller for WES data. Utilising a variant of the transformer architecture, the model is trained to call CNVs per exon.
- Berk Mandiracioglu
- , Furkan Ozden
- & A. Ercument Cicek
-
Article
| Open Access ACIDES: on-line monitoring of forward genetic screens for protein engineering
ACIDES: on-line monitoring of forward genetic screens for protein engineering
Screening mutated proteins is a versatile strategy in protein research, producing massive datasets when combined with NGS. Here, authors present ACIDES to estimate mutated protein fitness and aid protein engineering pipelines in a range of applications, including gene therapy.
- Takahiro Nemoto
- , Tommaso Ocari
- & Ulisse Ferrari
-
Article
| Open Access Pathway centric analysis for single-cell RNA-seq and spatial transcriptomics data with GSDensity
Pathway centric analysis for single-cell RNA-seq and spatial transcriptomics data with GSDensity
Clustering-based analysis has limited power in highly dynamic single-cell data, which is a common situation in tumour samples. Here, authors introduce GSDensity, enabling pathway-centric analysis for the direct integration of data with their domain knowledge.
- Qingnan Liang
- , Yuefan Huang
- & Ken Chen
-
Article
| Open Access Potent latency reversal by Tat RNA-containing nanoparticle enables multi-omic analysis of the HIV-1 reservoir
Potent latency reversal by Tat RNA-containing nanoparticle enables multi-omic analysis of the HIV-1 reservoir
Reactivating latent HIV reservoirs could be beneficial towards a functional cure. Here, the authors show that Tat-LNP effectively reactivates HIV while preserving the cell transcriptome. Upon reactivation, p24+ cells exhibit distinct genes and pathways potentially contributing to their persistence.
- Marion Pardons
- , Basiel Cole
- & Linos Vandekerckhove
-
Article
| Open Access Whole-genome sequencing reveals the molecular implications of the stepwise progression of lung adenocarcinoma
Whole-genome sequencing reveals the molecular implications of the stepwise progression of lung adenocarcinoma
Current sequencing technologies can shed light on the stepwise progression of lung adenocarcinoma. Here, the authors characterize tumor progression in lung adenocarcinomas from an early stage using short and long read whole-genome sequencing, bulk and spatial transcriptomics, and epigenomics.
- Yasuhiko Haga
- , Yoshitaka Sakamoto
- & Ayako Suzuki
-
Article
| Open Access A patterned human primitive heart organoid model generated by pluripotent stem cell self-organization
A patterned human primitive heart organoid model generated by pluripotent stem cell self-organization
Pluripotent stem cell-derived organoids can recapitulate significant hallmarks of human organ development and are becoming critical tools for human research. Here, the authors report significant technical steps for generating sophisticated synthetic human primitive heart organoids.
- Brett Volmert
- , Artem Kiselev
- & Aitor Aguirre
 Sex-specific developmental gene expression atlas unveils dimorphic gene networks in C. elegans
Sex-specific developmental gene expression atlas unveils dimorphic gene networks in C. elegans