
Article
|
Open Access
Featured
-
-
Article
| Open Access Deep mutational scanning reveals a correlation between degradation and toxicity of thousands of aspartoacylase variants
Deep mutational scanning reveals a correlation between degradation and toxicity of thousands of aspartoacylase variants
The details of how the protein folding and degradation systems collaborate to combat potentially toxic non-native proteins are unknown. Here the authors perform systematic studies of missense and nonsense variants of the cytosolic aspartoacylase, ASPA, where loss-of-function variants are linked to Canavan disease.
- Martin Grønbæk-Thygesen
- , Vasileios Voutsinos
- & Rasmus Hartmann-Petersen
-
Article
| Open Access Diphthamide deficiency promotes association of eEF2 with p53 to induce p21 expression and neural crest defects
Diphthamide deficiency promotes association of eEF2 with p53 to induce p21 expression and neural crest defects
Rare disease DEDSSH1-associated DPH1 mutations impair eEF2 diphthamide modification that leads to eEF2 as a transcriptional coactivator for p53 to enhance expression of the cell proliferation inhibitor p21, resulting in birth defects.
- Yu Shi
- , Daochao Huang
- & Weihong Song
-
Article
| Open Access Tracing genetic diversity captures the molecular basis of misfolding disease
Tracing genetic diversity captures the molecular basis of misfolding disease
Pei et al. applied Gaussian process-based machine learning to capture dynamic spatial covariance relationships managed by proteostasis to mediate cooperative folding on a residue basis as a standard model for precision disease management.
- Pei Zhao
- , Chao Wang
- & William E. Balch
-
Article
| Open Access Translation velocity determines the efficacy of engineered suppressor tRNAs on pathogenic nonsense mutations
Translation velocity determines the efficacy of engineered suppressor tRNAs on pathogenic nonsense mutations
An emerging therapeutic strategy is to suppress nonsense mutations with engineered suppressor tRNAs. Here, the authors show that the mRNA translation velocity is a key parameter determining the efficacy of suppressor tRNAs.
- Nikhil Bharti
- , Leonardo Santos
- & Zoya Ignatova
-
Article
| Open Access The aldehyde dehydrogenase 2 rs671 variant enhances amyloid β pathology
The aldehyde dehydrogenase 2 rs671 variant enhances amyloid β pathology
Here, Wang et al. report that the ALDH2 rs671 variant exacerbates amyloid-β pathology in the human brain. Mechanistically, the variant leads to 4-HNE accumulation, adducting Lys53 of C99 and promoting the production of Aβ40.
- Xia Wang
- , Jiayu Wang
- & Wei Ge
-
Article
| Open Access Biallelic NAA60 variants with impaired N-terminal acetylation capacity cause autosomal recessive primary familial brain calcifications
Biallelic NAA60 variants with impaired N-terminal acetylation capacity cause autosomal recessive primary familial brain calcifications
Most individuals with primary familial brain calcification (PFBC) remain genetically unsolved. Here the authors show that NAA60 biallelic variants cause PFBC, likely via reduced N-terminal acetylation and SLC20A2 levels with impaired phosphate uptake.
- Viorica Chelban
- , Henriette Aksnes
- & Henry Houlden
-
Article
| Open Access The evolutionary impact of childhood cancer on the human gene pool
The evolutionary impact of childhood cancer on the human gene pool
Pathogenic germline variants associated with childhood cancer risk could be subject to evolutionary constraints. Here, the authors analyse publicly available germline data in large cohorts and observe that paediatric cancer predisposition syndrome genes are highly constrained in the general population.
- Ulrik Kristoffer Stoltze
- , Jon Foss-Skiftesvik
- & Kjeld Schmiegelow
-
Article
| Open Access SNUPN deficiency causes a recessive muscular dystrophy due to RNA mis-splicing and ECM dysregulation
SNUPN deficiency causes a recessive muscular dystrophy due to RNA mis-splicing and ECM dysregulation
SNURPORTIN-1, encoded by the SNUPN gene, plays a key role in the nuclear import of spliceosomal small nuclear ribonucleoproteins, however its physiological function remains unclear. Here the authors report that recessive SNUPN mutations cause a distinct subtype of childhood muscular dystrophy and reveal SNURPORTIN-1’s role in muscle homeostasis, offering insights for new therapeutic strategies.
- Marwan Nashabat
- , Nasrinsadat Nabavizadeh
- & Nathalie Escande-Beillard
-
Article
| Open Access Cases of trisomy 21 and trisomy 18 among historic and prehistoric individuals discovered from ancient DNA
Cases of trisomy 21 and trisomy 18 among historic and prehistoric individuals discovered from ancient DNA
Information on the occurrence of aneuploidies in prehistory human populations are rare. Here, from a large screen of ancient human genomes and osteological examination, the authors find genetic evidence for six cases of trisomy 21 (Down syndrome) and one case of trisomy 18 (Edwards syndrome) in historic and prehistoric infants.
- Adam Benjamin Rohrlach
- , Maïté Rivollat
- & Kay Prüfer
-
Article
| Open Access Unraveling the genetic architecture of congenital vertebral malformation with reference to the developing spine
Unraveling the genetic architecture of congenital vertebral malformation with reference to the developing spine
Congenital vertebral malformation has a complex genetic architecture that isn’t fully understood. Here, the authors explore the genetic architecture of congenital vertebral malformation through case-control rare variant genetic analyses and embryonic transcriptome analyses of the developing spine.
- Sen Zhao
- , Hengqiang Zhao
- & Nan Wu
-
Article
| Open Access Large scale plasma proteomics identifies novel proteins and protein networks associated with heart failure development
Large scale plasma proteomics identifies novel proteins and protein networks associated with heart failure development
The pathobiology of heart failure (HF) is incompletely understood. The authors identify 37 circulating proteins and 5 protein modules associated with HF risk, with several demonstrating causal effects on HF, risk factors, or cardiac dysfunction by Mendelian randomization analysis.
- Amil M. Shah
- , Peder L. Myhre
- & Bing Yu
-
Article
| Open Access Fatty acid synthesis suppresses dietary polyunsaturated fatty acid use
Fatty acid synthesis suppresses dietary polyunsaturated fatty acid use
Polyunsaturated Fatty Acids (PUFA), such as omega-3 fatty acids, are recognized for their lipid lowering and anti-inflammatory properties. Here, the authors show that endogenous lipid synthesis controls the use of PUFA and thus determine the therapeutic benefit of omega-3 fatty acid supplementation.
- Anna Worthmann
- , Julius Ridder
- & Christian Schlein
-
Article
| Open Access vcfdist: accurately benchmarking phased small variant calls in human genomes
vcfdist: accurately benchmarking phased small variant calls in human genomes
Accurately benchmarking small variant calling accuracy is critical for the continued improvement of human genome sequencing. Here, the authors show that current approaches are biased towards certain variant representations and develop a new approach to ensure consistent and accurate benchmarking, regardless of the original variant representations.
- Tim Dunn
- & Satish Narayanasamy
-
Article
| Open Access SLC35D3 promotes white adipose tissue browning to ameliorate obesity by NOTCH signaling
SLC35D3 promotes white adipose tissue browning to ameliorate obesity by NOTCH signaling
White adipose tissue is closely associated with energy expenditure and obesity. Here, the authors show that SLC35D3 promotes white adipose tissue browning through the NOTCH1 signalling pathway and SLC35D3 may be a potential therapeutic target for obesity and related complications.
- Hongrui Wang
- , Liang Yu
- & Yibo Wang
-
Article
| Open Access Systematic analysis of paralogous regions in 41,755 exomes uncovers clinically relevant variation
Systematic analysis of paralogous regions in 41,755 exomes uncovers clinically relevant variation
Chameleolyser enables the accurate identification of genetic variants hidden within complex regions of the genome. Its application uncovers the disease-explanatory variant in 25 previously undiagnosed patients.
- Wouter Steyaert
- , Lonneke Haer-Wigman
- & Christian Gilissen
-
Article
| Open Access Simulation of undiagnosed patients with novel genetic conditions
Simulation of undiagnosed patients with novel genetic conditions
Rare Mendelian disorders pose a major diagnostic challenge, but evaluation of automated tools that aim to uncover causal genes tools is limited. Here, the authors present a computational pipeline that simulates realistic clinical datasets to address this deficit.
- Emily Alsentzer
- , Samuel G. Finlayson
- & Isaac S. Kohane
-
Article
| Open Access Genome-wide identification and phenotypic characterization of seizure-associated copy number variations in 741,075 individuals
Genome-wide identification and phenotypic characterization of seizure-associated copy number variations in 741,075 individuals
Here, the authors perform a meta-analysis in 26,699 people with seizures and 492,324 controls to identify 25 genome-wide significant copy-number variants. The discovered loci point to known disease genes and associations with clinical annotations.
- Ludovica Montanucci
- , David Lewis-Smith
- & Dennis Lal
-
Article
| Open Access Whole genome sequencing identifies genetic variants associated with neurogenic inflammation in rosacea
Whole genome sequencing identifies genetic variants associated with neurogenic inflammation in rosacea
Rosacea is a common, multi-factorial chronic inflammatory skin disorder. Here authors provide evidence of genetic predisposition by whole genome sequencing and whole exome sequencing of samples from familial cases, and by recapitulating a recurrent mutation in the LRRC4 gene in a mouse model, they find that neuron-derived vasoactive intestinal peptide is an important pathogenic factor for neurogenic inflammation in rosacea.’
- Zhili Deng
- , Mengting Chen
- & Ji Li
-
Article
| Open Access Deficit of homozygosity among 1.52 million individuals and genetic causes of recessive lethality
Deficit of homozygosity among 1.52 million individuals and genetic causes of recessive lethality
Genotypes causing pregnancy loss and perinatal mortality are depleted among living individuals and are therefore difficult to find. Here, using genetic data for 1.52 million individuals, the authors identify 25 genes with protein-altering variants exhibiting a strong deficit of homozygosity, suggesting they are essential for successful early development.
- Asmundur Oddsson
- , Patrick Sulem
- & Daniel F. Gudbjartsson
-
Article
| Open Access Variants in SART3 cause a spliceosomopathy characterised by failure of testis development and neuronal defects
Variants in SART3 cause a spliceosomopathy characterised by failure of testis development and neuronal defects
The SART3 gene encodes an RNA-binding protein critical for spliceosome function. Here, the authors find that bi-allelic variants in SART3 underlie a congenital condition characterised by neuro-developmental defects and 46,XY gonadal dysgenesis.
- Katie L. Ayers
- , Stefanie Eggers
- & Andrew H. Sinclair
-
Article
| Open Access Identifying high-impact variants and genes in exomes of Ashkenazi Jewish inflammatory bowel disease patients
Identifying high-impact variants and genes in exomes of Ashkenazi Jewish inflammatory bowel disease patients
Inflammatory bowel disease (IBD) is highly prevalent among the Ashkenazi Jewish population. Here, the authors identify novel IBD-associated variants and genes, validated by transcriptomic and phenome-wide associations.
- Yiming Wu
- , Kyle Gettler
- & Yuval Itan
-
Article
| Open Access DeMAG predicts the effects of variants in clinically actionable genes by integrating structural and evolutionary epistatic features
DeMAG predicts the effects of variants in clinically actionable genes by integrating structural and evolutionary epistatic features
Interpretation of rare genetic variants remains challenging. Here, the authors develop a supervised variant effect predictor for use in clinically actionable genes which incorporates evolutionary and structural relationships between residues and has balanced specificity and sensitivity.
- Federica Luppino
- , Ivan A. Adzhubei
- & Agnes Toth-Petroczy
-
Article
| Open Access Enhancer hijacking at the ARHGAP36 locus is associated with connective tissue to bone transformation
Enhancer hijacking at the ARHGAP36 locus is associated with connective tissue to bone transformation
Heterotopic ossification is a disorder characterized by the transformation of soft tissue to bone. In this study the authors report heterotopic ossification in a child caused by enhancer hijacking at the ARHGAP36 gene, the ectopic activation by an enhancer that it not its own.
- Uirá Souto Melo
- , Jerome Jatzlau
- & Malte Spielmann
-
Article
| Open Access A DNA tumor virus globally reprograms host 3D genome architecture to achieve immortal growth
A DNA tumor virus globally reprograms host 3D genome architecture to achieve immortal growth
The dynamic and temporal changes of host genome architecture during Epstein-Barr virus (EBV) transformation are not well known. Here the authors transform human primary B lymphocyte into lymphoblastoid cell lines (LCLs) with EBV and show that the host 3D genome is rewired to facilitate expression of key oncogenes.
- Chong Wang
- , Xiang Liu
- & Bo Zhao
-
Article
| Open Access A rare human variant that disrupts GPR10 signalling causes weight gain in mice
A rare human variant that disrupts GPR10 signalling causes weight gain in mice
The brain-expressed receptor GPR10 is involved in energy homeostasis in mice. Here the authors identify rare loss of function variants in GPR10 in people with severe obesity and showed that one of these variants causes obesity when modelled in mice, suggesting that future studies could explore GPR10 as a potential target for weight-loss therapy.
- Fleur Talbot
- , Claire H. Feetham
- & I. Sadaf Farooqi
-
Article
| Open Access Sequence terminus dependent PCR for site-specific mutation and modification detection
Sequence terminus dependent PCR for site-specific mutation and modification detection
Rapid and facile detection of specific nucleic acid modifications could have numerous applications. Here the authors present Specific Terminal Mediated Polymerase Chain Reaction (STEM-PCR) as a generic and accessible approach, and demonstrate proof-of-principle cancer biomarker detection.
- Gaolian Xu
- , Hao Yang
- & Hongchen Gu
-
Article
| Open Access TEFM variants impair mitochondrial transcription causing childhood-onset neurological disease
TEFM variants impair mitochondrial transcription causing childhood-onset neurological disease
Van Haute et al describe autosomal recessive TEFM variants that impair mitochondrial transcription elongation and reduce the levels of promoter distal mitochondrial RNA transcripts, leading to heterogeneous mitochondrial diseases with a treatable neuromuscular transmission defect.
- Lindsey Van Haute
- , Emily O’Connor
- & Rita Horvath
-
Article
| Open Access Personalized recurrence risk assessment following the birth of a child with a pathogenic de novo mutation
Personalized recurrence risk assessment following the birth of a child with a pathogenic de novo mutation
PREGCARE is a new strategy for families who had a child with a pathogenic de novo mutation, that efficiently identifies couples at higher recurrence risk due to parental mosaicism, while reassuring many others that their recurrence risk is negligible.
- Marie Bernkopf
- , Ummi B. Abdullah
- & Anne Goriely
-
Article
| Open Access Genome-wide screen of otosclerosis in population biobanks: 27 loci and shared associations with skeletal structure
Genome-wide screen of otosclerosis in population biobanks: 27 loci and shared associations with skeletal structure
Otosclerosis is a common form of hearing loss, with an unclear genetic etiology. Here, the authors perform a genome-wide association study meta-analysis of otosclerosis identifying 27 genetic loci, pointing to genes involved in bone remodeling, skeletal disorders and transforming growth factor β signaling.
- Joel T. Rämö
- , Tuomo Kiiskinen
- & Aarno Palotie
-
Article
| Open Access A gain-of-function TPC2 variant R210C increases affinity to PI(3,5)P2 and causes lysosome acidification and hypopigmentation
A gain-of-function TPC2 variant R210C increases affinity to PI(3,5)P2 and causes lysosome acidification and hypopigmentation
TPC2 is an important organellar Na+/Ca2+ release channel which regulates function of lysosomes and lysosome-related organelles. Here, Wang et al. demonstrate that a gain-of-function mutation (R210C) in TPC2 leads to hypopigmentaion, enlarged endolysosomes, enhanced lysosomal Ca2+ release and hyper-acidification.
- Qiaochu Wang
- , Zengge Wang
- & Wei Li
-
Article
| Open Access PEAC-seq adopts Prime Editor to detect CRISPR off-target and DNA translocation
PEAC-seq adopts Prime Editor to detect CRISPR off-target and DNA translocation
It is still a challenge to accurately identify off-target endonuclease edits. Here the authors report PEAC-seq using a Prime Editor to insert a tag to the editing sites and enrich the tagged regions with site-specific primers for sequencing: they show that PEAC-seq could identify DNA translocations.
- Zhenxing Yu
- , Zhike Lu
- & Lijia Ma
-
Article
| Open Access Automated high-throughput genome editing platform with an AI learning in situ prediction model
Automated high-throughput genome editing platform with an AI learning in situ prediction model
A large number of cell disease models with pathogenic SNVs are needed. Here the authors report an automated high-throughput platform to perform the genome editing process from gRNA design to the analysis of the editing results; they characterise in situ base editing outcomes.
- Siwei Li
- , Jingjing An
- & Meng Wang
-
Article
| Open Access Cholesterol-induced LRP3 downregulation promotes cartilage degeneration in osteoarthritis by targeting Syndecan-4
Cholesterol-induced LRP3 downregulation promotes cartilage degeneration in osteoarthritis by targeting Syndecan-4
This study demonstrates a role of cholesterol metabolism-related gene, Lrp3, in cartilage degeneration and osteoarthritis pathogenesis. LRP3 positively regulates cartilage extracellular matrix metabolism by targeting syndecan-4 via Ras signalling, implicating the cholesterol-LRP3-SDC4 axis in osteoarthritic cartilage degeneration.
- Chenxi Cao
- , Yuanyuan Shi
- & Yingfang Ao
-
Article
| Open Access Dominant ARF3 variants disrupt Golgi integrity and cause a neurodevelopmental disorder recapitulated in zebrafish
Dominant ARF3 variants disrupt Golgi integrity and cause a neurodevelopmental disorder recapitulated in zebrafish
Disruptions to the ER-Golgi network can lead to neurodevelopmental disorders, though our understanding of these Golgipathies remains incomplete. Here Lauri, Tartaglia and colleagues show that ARF3 mutations cause a rare pediatric neurological disorder and perform detailed molecular characterization in fish.
- Giulia Fasano
- , Valentina Muto
- & Marco Tartaglia
-
Article
| Open Access Specialist multidisciplinary input maximises rare disease diagnoses from whole genome sequencing
Specialist multidisciplinary input maximises rare disease diagnoses from whole genome sequencing
Whole genome sequencing is emerging as a first-line test for rare genetic diseases. In this study, authors maximise diagnoses by supplementing existing semiautomated analyses with clinically driven reevaluation of genomic data by a specialist multidisciplinary team.
- William L. Macken
- , Micol Falabella
- & Robert D. S. Pitceathly
-
Article
| Open Access Analysis of clinically relevant variants from ancestrally diverse Asian genomes
Analysis of clinically relevant variants from ancestrally diverse Asian genomes
Clinically significant genetic variation in Asian populations is under-characterized. Here, the authors show the diversity in prevalence and spectrum of human disease and pharmacogenetic variants in a multi-ethnic Asian population.
- Sock Hoai Chan
- , Yasmin Bylstra
- & Weng Khong Lim
-
Article
| Open Access Systematic Mendelian randomization using the human plasma proteome to discover potential therapeutic targets for stroke
Systematic Mendelian randomization using the human plasma proteome to discover potential therapeutic targets for stroke
Mendelian randomization can be used to mimic the effects of protein-targeting drugs in a population of individuals. Here, the authors have identified potential causal proteins for stroke in a two-sample Mendelian randomization framework, providing potential stroke therapeutic targets.
- Lingyan Chen
- , James E. Peters
- & Joanna M. M. Howson
-
Article
| Open Access The mutational signatures of formalin fixation on the human genome
The mutational signatures of formalin fixation on the human genome
Many archived tumour samples are stored as formalin-fixed and paraffin-embedded (FFPE) blocks, but this treatment can impact downstream genomics analyses. Here, the authors derive the mutational signatures of formalin on the cancer genome, and present FFPEsig, an algorithm that can distinguish and correct FFPE mutational signatures in archived cancer samples.
- Qingli Guo
- , Eszter Lakatos
- & Ville Mustonen
-
Article
| Open Access Endophenotype effect sizes support variant pathogenicity in monogenic disease susceptibility genes
Endophenotype effect sizes support variant pathogenicity in monogenic disease susceptibility genes
Accurate classification of genetic variants is critical for research and patient care. Here, the authors report that population-based associations between rare variants and quantitative endophenotypes for monogenic diseases can provide support for variant pathogenicity.
- Jennifer L. Halford
- , Valerie N. Morrill
- & Steven A. Lubitz
-
Article
| Open Access Comprehensive genomic and epigenomic analysis in cancer of unknown primary guides molecularly-informed therapies despite heterogeneity
Comprehensive genomic and epigenomic analysis in cancer of unknown primary guides molecularly-informed therapies despite heterogeneity
The identification of molecular biomarkers in cancer of unknown primary site (CUP) cases may enable the improvement of prognosis in these patients. Here, the authors integrate whole genome/exome, transcriptome and methylome data in 70 CUP patients, recommend therapies based on their analysis and report clinical outcome data.
- Lino Möhrmann
- , Maximilian Werner
- & Hanno Glimm
-
Article
| Open Access An automated 13.5 hour system for scalable diagnosis and acute management guidance for genetic diseases
An automated 13.5 hour system for scalable diagnosis and acute management guidance for genetic diseases
Rapid diagnosis and implementation of treatments is crucial in many genetic conditions. Here the authors describe Genome-to-Treatment, a virtual disease management system that can achieve a rapid diagnosis by expedited whole genome sequencing in 13.5 hours and provide guidance to clinicians for possible therapies.
- Mallory J. Owen
- , Sebastien Lefebvre
- & Stephen F. Kingsmore
-
Article
| Open Access The contribution of common regulatory and protein-coding TYR variants to the genetic architecture of albinism
The contribution of common regulatory and protein-coding TYR variants to the genetic architecture of albinism
Albinism is a rare disorder often caused by high-effect rare variants in the TYR gene. Here, the authors study a large albinism cohort and find that a common variant in the TYR promoter contributes to albinism by modifying the penetrance of other common variants, demonstrating a complex genetic architecture.
- Vincent Michaud
- , Eulalie Lasseaux
- & Panagiotis I. Sergouniotis
-
Article
| Open Access Clonal reconstruction from co-occurrence of vector integration sites accurately quantifies expanding clones in vivo
Clonal reconstruction from co-occurrence of vector integration sites accurately quantifies expanding clones in vivo
High transduction rates of viral vectors ensure good gene delivery; however multiple integration events can occur in the same cell. Here the authors use correlations between repeated measurements of integration site abundances to estimate their mutual similarity and identify clusters of co-occurring sites.
- Sebastian Wagner
- , Christoph Baldow
- & Ingmar Glauche
-
Article
| Open Access Common genetic variation associated with Mendelian disease severity revealed through cryptic phenotype analysis
Common genetic variation associated with Mendelian disease severity revealed through cryptic phenotype analysis
The severity of rare genetic diseases often varies between individuals, but small sample sizes make it difficult to identify contributing factors. Here, the authors use biobank-scale clinical and genetic data to investigate a role for common genetic variation.
- David R. Blair
- , Thomas J. Hoffmann
- & Joseph T. Shieh
-
Article
| Open Access The chromatin remodeller ATRX facilitates diverse nuclear processes, in a stochastic manner, in both heterochromatin and euchromatin
The chromatin remodeller ATRX facilitates diverse nuclear processes, in a stochastic manner, in both heterochromatin and euchromatin
The chromatin remodeling complex ATRX can promote gene expression, for example by binding G-quadruplexes (G4s) to prevent their negative effect on expression. Here the authors use a single-cell approach to show that only a subset of erythroid cells isolated from patients with ATRX mutations have reduced chromatin accessibility and alpha globin expression, suggesting a stochastic process.
- Julia Truch
- , Damien J. Downes
- & Richard J. Gibbons
-
Article
| Open Access Variants in ASPH cause exertional heat illness and are associated with malignant hyperthermia susceptibility
Variants in ASPH cause exertional heat illness and are associated with malignant hyperthermia susceptibility
The genetic cause(s) of malignant hyperthermia and exertional heat illness are unknown in approximately 30% of cases. To address this barrier, the authors performed genome sequencing on a large cohort of cases, identifying rare variants in ASPH, a gene encoding junctin, and validating them in animal and cell models.
- Yukari Endo
- , Linda Groom
- & James J. Dowling
-
Article
| Open Access Neonatal gene therapy achieves sustained disease rescue of maple syrup urine disease in mice
Neonatal gene therapy achieves sustained disease rescue of maple syrup urine disease in mice
Maple syrup urine disease (MSUD) is a rare inborn error of metabolism, which is currently treated with life-long low-protein diet that can be challenging to maintain. Here the authors develop an AAV8-directed gene therapy providing sustainable disease rescue in a mouse model of MSUD.
- Clément Pontoizeau
- , Marcelo Simon-Sola
- & Manuel Schiff
-
Article
| Open Access Establishment of mouse model of inherited PIGO deficiency and therapeutic potential of AAV-based gene therapy
Establishment of mouse model of inherited PIGO deficiency and therapeutic potential of AAV-based gene therapy
Inherited GPI deficiency (IGD) is caused by PIGO mutations. Here, the authors generate a mouse model of IGD and show that AAV-mediate gene therapy, for knock-in as well as extra-chromosomal expression of Pigo cDNA, ameliorates pathology in the mice.
- Ryoko Kuwayama
- , Keiichiro Suzuki
- & Yoshiko Murakami
-
Article
| Open Access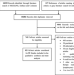 A systematic review and meta-analysis of gene therapy with hematopoietic stem and progenitor cells for monogenic disorders
A systematic review and meta-analysis of gene therapy with hematopoietic stem and progenitor cells for monogenic disorders
Ex-vivo gene therapy with hematopoietic stem and progenitor cells (HSPCs) is a promising treatment for monogenic diseases. Here the authors report a systematic review and meta-analysis of available evidence assessing clinical outcomes of HSPC gene therapy from clinical trials.
- Francesca Tucci
- , Stefania Galimberti
- & Alessandro Aiuti
 Ciliary tip actin dynamics regulate photoreceptor outer segment integrity
Ciliary tip actin dynamics regulate photoreceptor outer segment integrity