
Abstract
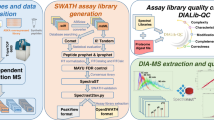
Technological advances in mass spectrometry and proteomics have made it possible to perform larger-scale and more-complex experiments. The volume and complexity of the resulting data create major challenges for downstream analysis. In particular, next-generation data-independent acquisition (DIA) experiments enable wider proteome coverage than more traditional targeted approaches but require computational workflows that can manage much larger datasets and identify peptide sequences from complex and overlapping spectral features. Data-processing tools such as FragPipe, DIA-NN and Spectronaut have undergone substantial improvements to process spectral features in a reasonable time. Statistical analysis tools are needed to draw meaningful comparisons between experimental samples, but these tools were also originally designed with smaller datasets in mind. This protocol describes an updated version of MSstats that has been adapted to be compatible with large-scale DIA experiments. A very large DIA experiment, processed with FragPipe, is used as an example to demonstrate different MSstats workflows. The choice of workflow depends on the user’s computational resources. For datasets that are too large to fit into a standard computer’s memory, we demonstrate the use of MSstatsBig, a companion R package to MSstats. The protocol also highlights key decisions that have a major effect on both the results and the processing time of the analysis. The MSstats processing can be expected to take 1–3 h depending on the usage of MSstatsBig. The protocol can be run in the point-and-click graphical user interface MSstatsShiny or implemented with minimal coding expertise in R.
Key points
-
Technological advances in bottom-up mass spectrometry-based proteomics have resulted in a substantial increase in the volume and complexity of the resulting data, and for comparative studies, large numbers of samples are required to get statistically meaningful results.
-
MSstats can be used to perform statistical analysis of the data after the peptides and proteins have been identified and quantified. MSstatsBig is a variant specifically designed to manage very large datasets.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others


Data availability
The dataset used in this protocol is freely available at https://pdc.cancer.gov/pdc/study/PDC000200 and MassIVE (https://massive.ucsd.edu/ProteoSAFe/dataset.jsp?task=6847574c13964a1f9482ee4d71f33eb1. The quantification results from FragPipe and the MSstats processed data at each step are available in the MassIVE.quant Reanalysis RMSV000000696.1. Source data are provided with this paper.
Code availability
All analysis scripts to recreate Procedure 2 can be found in the same MassIVE.quant Reanalysis RMSV000000696.1.
References
Shuken, S. R. An introduction to mass spectrometry-based proteomics. J. Proteom. Res. 22, 2151–2171 (2023).
Aebersold, R. & Mann, M. Mass spectrometry-based proteomics. Nature 422, 198–207 (2003).
Ong, S.-E. & Mann, M. Mass spectrometry–based proteomics turns quantitative. Nat. Chem. Biol. 1, 252–262 (2005).
Borràs, E. & Sabidó, E. What is targeted proteomics? A concise revision of targeted acquisition and targeted data analysis in mass spectrometry. Proteomics 17, 1700180 (2017).
Mann, M. & Jensen, O. N. Proteomic analysis of post-translational modifications. Nat. Biotechnol. 21, 255–261 (2003).
Li, Z. et al. Systematic comparison of label-free, metabolic labeling, and isobaric chemical labeling for quantitative proteomics on LTQ Orbitrap Velos. J. Proteome Res. 11, 1582–1590 (2012).
Poulos, R. C. et al. Strategies to enable large-scale proteomics for reproducible research. Nat. Commun. 11, 3793 (2020).
Cai, X. et al. PulseDIA: data-independent acquisition mass spectrometry using multi-injection pulsed gas-phase fractionation. J. Proteome Res. 20, 279–288 (2021).
Krzywinski, M. & Altman, N. Power and sample size. Nat. Methods 10, 1139–1140 (2013).
Kong, A. T., Leprevost, F. V., Avtonomov, D. M., Mellacheruvu, D. & Nesvizhskii, A. I. MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry–based proteomics. Nat. Methods 14, 513–520 (2017).
Demichev, V., Messner, C. B., Vernardis, S. I., Lilley, K. S. & Ralser, M. DIA-NN: neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods 17, 41–44 (2020).
Bernhardt, O. M. et al. Spectronaut: A Fast and Efficient Algorithm for MRM-Like Processing of Data Independent Acquisition (SWATH-MS) Data. Presented at Proceedings of the 60th ASMS Conference on Mass Spectrometry and Allied Topics, Vancouver, BC, Canada, (unpublished), https://f1000research.com/posters/1096450 (2012).
MacLean, B. et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 (2010).
Tyanova, S., Temu, T. & Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 11, 2301–2319 (2016).
Röst, H. L. et al. OpenSWATH enables automated, targeted analysis of data-independent acquisition MS data. Nat. Biotechnol. 32, 219–223 (2014).
Zhang, F., Ge, W., Ruan, G., Cai, X. & Guo, T. Data-independent acquisition mass spectrometry-based proteomics and software tools: a glimpse in 2020. Proteomics 20, e1900276 (2020).
Demichev, V. et al. dia-PASEF data analysis using FragPipe and DIA-NN for deep proteomics of low sample amounts. Nat. Commun. 13, 3944 (2022).
Yu, F. et al. Analysis of DIA proteomics data using MSFragger-DIA and FragPipe computational platform. Nat. Commun. 14, 4154 (2023).
Käll, L. & Vitek, O. Computational mass spectrometry–based proteomics. PLoS Comput. Biol. 7, e1002277 (2011).
Molloy, M. P., Brzezinski, E. E., Hang, J., McDowell, M. T. & VanBogelen, R. A. Overcoming technical variation and biological variation in quantitative proteomics. Proteomics 3, 1912–1919 (2003).
Clough, T., Thaminy, S., Ragg, S., Ruedi, A. & Vitek, O. Statistical protein quantification and significance analysis in label-free LC-MS experiments with complex designs. BMC Bioinforma. 13, S6 (2012).
Tsai, T.-H. et al. Selection of features with consistent profiles improves relative protein quantification in mass spectrometry experiments. Mol. Cell. Proteom. 19, 944–959 (2020).
Girden, E. R. ANOVA: Repeated Measures (Sage Publications, 1992).
Kohler, D. et al. MSstats version 4.0: statistical analyses of quantitative mass spectrometry-based proteomic experiments with chromatography-based quantificationat scale. J. Proteome Res. 22, 1466–1482 (2023).
Choi, M. et al. MSstats: an R package for statistical analysis of quantitative mass spectrometry-based proteomic experiments. Bioinformatics 30, 2524–2526 (2014).
Goeminne, L. J. E., Sticker, A., Martens, L., Gevaert, K. & Clement, L. MSqRob takes the missing hurdle: uniting intensity- and count-based proteomics. Anal. Chem. 92, 6278–6287 (2020).
Sticker, A., Goeminne, L., Martens, L. & Clement, L. Robust summarization and inference in proteomewide label-free quantification. Mol. Cell. Proteom. 19, 1209–1219 (2020).
Goeminne, L. J. E., Gevaert, K. & Clement, L. Peptide-level robust ridge regression improves estimation, sensitivity, and specificity in data-dependent quantitative label-free shotgun proteomics. Mol. Cell. Proteom. 15, 657–668 (2016).
Zhu, et al. DEqMS: a method for accurate variance estimation in differential protein expression analysis. Mol. Cell. Proteom. 19, 1047–1057 (2020).
Wolski, W. E. et al. prolfqua: a comprehensive R-package for proteomics differential expression analysis. J. Proteome Res. 22, 1092–1104 (2023).
Bai, M. et al. LFQ-based peptide and protein intensity differential expression analysis. J. Proteome Res. 22, 2114–2123 (2023).
Gatto, L. & Vanderaa, C. R Package Version 1.13.1, https://github.com/RforMassSpectrometry/QFeatures (2023).
Simmons, J. P., Nelson, L. D. & Simonsohn, U. False-positive psychology: undisclosed flexibility in data collection and analysis allows presenting anything as significant. Psychol. Sci. 22, 1359–1366 (2011).
Kohler, D. et al. MSstatsShiny: a GUI for versatile, scalable, and reproducible statistical analyses of quantitative proteomic experiments. J. Proteome Res. 22, 551–556 (2023).
Yang, K. L. et al. MSBooster: improving peptide identification rates using deep learning-based features. Nat. Commun. 14, 4539 (2023).
Leprevost, F. D. V. et al. Philosopher: a versatile toolkit for shotgun proteomics data analysis. Nat. Methods 17, 869–870 (2020).
Kohler, D. et al. MSstatsPTM: statistical relative quantification of posttranslational modifications in bottom-up mass spectrometry-based proteomics. Mol. Cell. Proteom. 22, 100477 (2023).
Huang, T. et al. MSstatsTMT: statistical detection of differentially abundant proteins in experiments with isobaric labeling and multiple mixtures. Mol. Cell. Proteom. 19, 1706–1723 (2020).
Malinovska, L. et al. Proteome-wide structural changes measured with limited proteolysis-mass spectrometry: an advanced protocol for high-throughput applications. Nat. Protoc. 18, 659–682 (2022).
Richardson, N., et al. Apache/Arrow, https://github.com/apache/arrow/, https://arrow.apache.org/docs/r/ (2023).
Zaharia, M., Xin, R. S., Wendell, P., Das, T. & Armbrust, M. Apache Spark: a unified engine for big data processing. Commun. ACM 59, 56–65 (2016).
Feng, et al. Global analysis of protein structural changes in complex proteomes. Nat. Biotechnol. 32, 1036–1044 (2014).
Clark, D. J., Dhanasekaran, S. M., Petralia, F., Wang, P. & Zhang, H. Integrated proteogenomic characterization of clear cell renal cell carcinoma. Cell 179, 964–983 (2019).
Dowle, M. & Srinivasan, A. data.table, https://r-datatable.com, https://Rdatatable.gitlab.io/data.table, https://github.com/Rdatatable/data.table (2023).
Venables, W. & Ripley, B. Modern Applied Statistics with S 359–364 (Springer, 2002).
Acknowledgements
We thank J. Carver for his help in setting up the MassIVE container that allowed us to share the datasets and analysis code for this paper. This work was supported by awards NSF-BIO/DBI-1759736 (to O.V.), NSF-BIO/DBI-1950412 (to O.V.) and NIH-NLM-1R01LM013115 (to O.V.), the Chan-Zuckerberg Foundation (to O.V.) and National Institutes of Health grants R01-GM-094231 and U24-CA271037 (to A.I.N.). M.S. was partially financially supported by the National Science Centre, Poland, grant Preludium 2020/37/N/ST6/04070.
Author information
Authors and Affiliations
Contributions
D.K. analyzed the data in MSstats and wrote the relevant MSstats sections of the manuscript. D.K. and O.V. wrote the introduction for the manuscript. M.S. implemented the methods in MSstatsBig. F.Y. analyzed the data by using FragPipe and wrote the relevant FragPipe sections of the paper. F.Y. and A.I.N. determined the experimental dataset for the manuscript. A.I.N., O.V. and D.K. conceptually developed and scoped the manuscript. All authors provided feedback and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
A.I.N. and F.Y. receive royalties from the University of Michigan for the sale of MSFragger and IonQuant software licenses to commercial entities. All license transactions are managed by the University of Michigan Innovation Partnerships office, and all proceeds are subject to the university technology transfer policy. The other authors declare no competing interests.
Peer review
Peer review information
Nature Protocols thanks Chu Wang, Witold Wolski and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
Key references using this protocol
Kohler, D. et al. J. Proteome Res. 22, 1466–1482 (2023)
Kohler, D. et al. J. Proteome Res. 22, 551–556 (2023)
Kong, A. et al. Nat. Methods 14, 513–520 (2017)
Yu, F. et al. Nat. Commun. 14, 4154 (2023)
Clark, D. J. et al. Cell 179, 964–983 (2019)
Extended data
Supplementary information
Supplementary Information
Supplementary Methods 1 and 2, Figs. 1–9 and Tables 1 and 2
Source data
Source Data Fig. 2
Statistical source data for Fig. 2b–d
Source Data Fig. 3
Statistical source data for Fig. 3a–c
Source Data Fig. 4
Statistical source data for Fig. 4
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kohler, D., Staniak, M., Yu, F. et al. An MSstats workflow for detecting differentially abundant proteins in large-scale data-independent acquisition mass spectrometry experiments with FragPipe processing. Nat Protoc (2024). https://doi.org/10.1038/s41596-024-01000-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41596-024-01000-3
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
