
Abstract
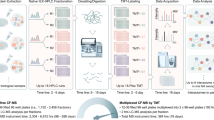
Affinity purification coupled to mass spectrometry (AP–MS) is the method of choice for analyzing protein–protein interactions, but common protocols frequently recover only the most stable interactions and tend to result in low bait yield for membrane proteins. Here, we present a novel, deep interactome sequencing approach called CoPIT (co-interacting protein identification technology), which allows comprehensive identification and analysis of membrane protein interactomes and their dynamics. CoPIT integrates experimental and computational methods for a coimmunoprecipitation (Co-IP)-based workflow from sample preparation for mass spectrometric analysis to visualization of protein–protein interaction networks. The approach particularly improves the results for membrane protein interactomes, which have proven to be difficult to identify and analyze. CoPIT was used successfully to identify the interactome of the cystic fibrosis transmembrane conductance regulator (CFTR), demonstrating its validity and performance. The experimental step in this case achieved up to 100-fold-higher bait yield than previous methods by optimizing lysis, elution, sample clean-up and detection of interacting proteins by multidimensional protein identification technology (MudPIT). Here, we further provide evidence that CoPIT is applicable to other types of proteins as well, and that it can be successfully used as a general Co-IP method. The protocol describes all steps, ranging from considerations for experimental design, Co-IP, preparation of the sample for mass spectrometric analysis, and data analysis steps, to the final visualization of interaction networks. Although the experimental part can be performed in <3 d, data analysis may take up to a few weeks.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others


Change history
30 November 2017
In the version of this article initially published, Steps 11 and 20 mentioned an incorrect reagent. The correct text is: "Immediately add 1.5 ml of ice-cold TNI lysis buffer per 15-cm cell culture dish” (Step 11) and "Remove the supernatant, transfer the beads to a new tube and wash the beads three times with 20–100 bead volumes of TNI lysis buffer" (Step 20). The TNI lysis buffer was also not listed in the Materials. These errors have been corrected, and the following text was added to the Materials section: "TNI lysis buffer TNI lysis buffer is 0.5% Igepal CA-630, 50 mM Tris (pH 7.5), 250 mM NaCl, 1 mM EDTA, 1× Complete ULTRA EDTA-free Protease Inhibitor cocktail and 1× PhosStop." These errors, made during production, have been corrected in the HTML and PDF versions of the article.
References
Gingras, A.C., Gstaiger, M., Raught, B. & Aebersold, R. Analysis of protein complexes using mass spectrometry. Nat. Rev. Mol. Cell Biol. 8, 645–654 (2007).
Cravatt, B.F., Simon, G.M. & Yates, J.R. III. The biological impact of mass-spectrometry-based proteomics. Nature 450, 991–1000 (2007).
Russ, A.P. & Lampel, S. The druggable genome: an update. Drug Discov. Today 10, 1607–1610 (2005).
Hopkins, A.L. & Groom, C.R. The druggable genome. Nat. Rev. Drug Discov. 1, 727–730 (2002).
Wallin, E. & von Heijne, G. Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Protein Sci. 7, 1029–1038 (1998).
Almen, M.S., Nordstrom, K.J., Fredriksson, R. & Schioth, H.B. Mapping the human membrane proteome: a majority of the human membrane proteins can be classified according to function and evolutionary origin. BMC Biol. 7, 50 (2009).
Blonder, J. et al. Enrichment of integral membrane proteins for proteomic analysis using liquid chromatography-tandem mass spectrometry. J. Proteome Res. 1, 351–360 (2002).
Howell, K.E. & Palade, G.E. Hepatic Golgi fractions resolved into membrane and content subfractions. J. Cell Biol. 92, 822–832 (1982).
Wu, C.C., MacCoss, M.J., Howell, K.E. & Yates, J.R. III. A method for the comprehensive proteomic analysis of membrane proteins. Nat. Biotechnol. 21, 532–538 (2003).
Santoni, V., Kieffer, S., Desclaux, D., Masson, F. & Rabilloud, T. Membrane proteomics: use of additive main effects with multiplicative interaction model to classify plasma membrane proteins according to their solubility and electrophoretic properties. Electrophoresis 21, 3329–3344 (2000).
Mitra, S.K., Gantt, J.A., Ruby, J.F., Clouse, S.D. & Goshe, M.B. Membrane proteomic analysis of Arabidopsis thaliana using alternative solubilization techniques. J. Proteome Res. 6, 1933–1950 (2007).
Macher, B.A. & Yen, T.Y. Proteins at membrane surfaces-a review of approaches. Mol. Biosyst. 3, 705–713 (2007).
Washburn, M.P., Wolters, D. & Yates, J.R. III. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 19, 242–247 (2001).
Palade, G. Intracellular aspects of the process of protein synthesis. Science 189, 347–358 (1975).
Wickner, W. & Schekman, R. Protein translocation across biological membranes. Science 310, 1452–1456 (2005).
Rapoport, T.A. Protein translocation across the eukaryotic endoplasmic reticulum and bacterial plasma membranes. Nature 450, 663–669 (2007).
Moore, I. & Murphy, A. Validating the location of fluorescent protein fusions in the endomembrane system. Plant Cell 21, 1632–1636 (2009).
Sastry, M.S., Zhou, W. & Baneyx, F. Integrity of N- and C-termini is important for E. coli Hsp31 chaperone activity. Protein Sci. 18, 1439–1447 (2009).
Choi, H. et al. SAINT: probabilistic scoring of affinity purification-mass spectrometry data. Nat. Methods 8, 70–73 (2011).
Breitkreutz, A. et al. A global protein kinase and phosphatase interaction network in yeast. Science 328, 1043–1046 (2010).
Sowa, M.E., Bennett, E.J., Gygi, S.P. & Harper, J.W. Defining the human deubiquitinating enzyme interaction landscape. Cell 138, 389–403 (2009).
Jager, S. et al. Global landscape of HIV-human protein complexes. Nature 481, 365–370 (2012).
Pankow, S., Bamberger, C., Calzolari, D., Bamberger, A. & Yates, J.R. Characterization of membrane protein interactomes by Co-interacting Protein Identification Technology (CoPIT). Protoc. Exch. (2015).
Pankow, S. et al. F508 CFTR interactome remodelling promotes rescue of cystic fibrosis. Nature 528, 510–516 (2015).
Zielenski, J. et al. Genomic DNA sequence of the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Genomics 10, 214–228 (1991).
Rommens, J.M. et al. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science 245, 1059–1065 (1989).
Sheppard, D.N. & Welsh, M.J. Structure and function of the CFTR chloride channel. Physiol. Rev. 79, S23–S45 (1999).
Collins, F.S. Cystic fibrosis: molecular biology and therapeutic implications. Science 256, 774–779 (1992).
Jensen, T.J. et al. Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell 83, 129–135 (1995).
Lukacs, G.L. et al. The delta F508 mutation decreases the stability of cystic fibrosis transmembrane conductance regulator in the plasma membrane. Determination of functional half-lives on transfected cells. J. Biol. Chem. 268, 21592–21598 (1993).
Abelin, J.G. et al. Complementary IMAC enrichment methods for HLA-associated phosphopeptide identification by mass spectrometry. Nat. Protoc. 10, 1308–1318 (2015).
Schieltz, D.M., Washburn, M.P. & Hays, L.G. Analysis of complex protein mixtures using nano-LC coupled to MS/MS. CSH Protoc. 2006, (5); http://dx.doi.org/10.1101/pdb.prot4553 (2006).
Wolters, D.A., Washburn, M.P. & Yates, J.R. III. An automated multidimensional protein identification technology for shotgun proteomics. Anal. Chem. 73, 5683–5690 (2001).
Schieltz, D.M. & Washburn, M.P. Analysis of complex protein mixtures using Multidimensional Protein Identification Technology (MuDPIT). CSH Protoc. 2006, (5); http://dx.doi.org/10.1101/pdb.prot4553 (2006).
Li, X.J., Zhang, H., Ranish, J.A. & Aebersold, R. Automated statistical analysis of protein abundance ratios from data generated by stable-isotope dilution and tandem mass spectrometry. Anal. Chem. 75, 6648–6657 (2003).
Ranish, J.A. et al. Identification of TFB5, a new component of general transcription and DNA repair factor IIH. Nat. Genet. 36, 707–713 (2004).
Smoot, M.E., Ono, K., Ruscheinski, J., Wang, P.L. & Ideker, T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics 27, 431–432 (2011).
Montojo, J. et al. GeneMANIA Cytoscape plugin: fast gene function predictions on the desktop. Bioinformatics 26, 2927–2928 (2010).
Morris, J.H. et al. Affinity purification-mass spectrometry and network analysis to understand protein-protein interactions. Nat. Protoc. 9, 2539–2554 (2014).
Pavlopoulos, G.A., Hooper, S.D., Sifrim, A., Schneider, R. & Aerts, J. Medusa: a tool for exploring and clustering biological networks. BMC Res. Notes 4, 384 (2011).
Wang, X. et al. Hsp90 cochaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis. Cell 127, 803–815 (2006).
Acknowledgements
We thank J. Clancy (University of Alabama, Birmingham, AL) for kindly providing the cell lines used in this work. We thank D. Cociorva and T. Xu for suggestions and comments on data analysis strategies, Y.L. Tan from J. Kelly's laboratory for performing the GC IPs and N. Huang from A. Rao's laboratory for performing the Tet2 IPs. This work was supported by NIH grants 5R01HL079442 (J.R.Y.), P01AG031097 (J.R.Y.), P41 RR011823 (J.R.Y.), DK075295 (J.W.K.) and HHSN268201000035C (J.R.Y.), and CFF mass spectrometry fellowship BALCH050X6 (S.P. and J.R.Y.).
Author information
Authors and Affiliations
Contributions
S.P. and C.B. developed experimental methods and performed experiments, data analysis and mass spectrometric measurements. C.B., A.B. and S.P. developed the statistical analysis. D.C. and C.B. developed the Radial Topology Viewer. J.R.Y., S.P. and C.B. designed the study and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Integrated supplementary information
Supplementary Figure 1 Sequence coverage achieved by CoPIT.
A. Sequence coverage of SMG1, GC and Tet2 indicating identified peptides (green). B. Sequence coverage of Calnexin indicating identified peptides (yellow), transmembrane domain (green) and cleaved signal peptide (turquoise).
Supplementary Figure 2 Recovery of hydrophobic CFTR peptides by MudPIT.
High salt concentrations are necessary for elution of hydrophobic CFTR peptides from SCX. The graph shows the distribution of Kyte-Dolittle (KD) values indicating hydrophobicity of identified ∆F508 CFTR peptides over the different MudPIT steps and corresponding salt concentrations. Error bars indicate mean with s.e.m.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1 and 2 (PDF 576 kb)
Rights and permissions
About this article

Cite this article
Pankow, S., Bamberger, C., Calzolari, D. et al. Deep interactome profiling of membrane proteins by co-interacting protein identification technology. Nat Protoc 11, 2515–2528 (2016). https://doi.org/10.1038/nprot.2016.140
Published:
Issue Date:
DOI: https://doi.org/10.1038/nprot.2016.140
This article is cited by
-
Targeted quantitation of CFTR protein expression in vivo using immunoprecipitation & parallel reaction monitoring tandem mass spectrometry
Translational Medicine Communications (2022)
-
SFPQ rescues F508del-CFTR expression and function in cystic fibrosis bronchial epithelial cells
Scientific Reports (2021)
-
Thy-1 depletion and integrin β3 upregulation-mediated PI3K-Akt-mTOR pathway activation inhibits lung fibroblast autophagy in lipopolysaccharide-induced pulmonary fibrosis
Laboratory Investigation (2019)
-
Heterologous calcium-dependent inactivation of Orai1 by neighboring TRPV1 channels modulates cell migration and wound healing
Communications Biology (2019)
-
Deducing the presence of proteins and proteoforms in quantitative proteomics
Nature Communications (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
