
Abstract
Here we present four unrelated families with six individuals that have infantile-onset developmental delay/regression and epilepsy. Whole-exome sequencing revealed compound heterozygous mutations, c.[283G>A];[607G>A] in a gene encoding prolyl-tRNA synthetase (PARS2) in one family. Two pairs of compound heterozygous mutations, c.[151C>T];[1184T>G] and c.[707T>G];[594+1G>A], and a homozygous mutation, c.[500A>G];[500A>G], in a gene encoding asparaginyl-tRNA synthetase (NARS2) were also identified in the other three families. Mutations in genes encoding aminoacyl-tRNA synthetases cause gene-specific mitochondrial disorders. Biallelic PARS2 or NARS2 mutations are reported to cause Alpers’ syndrome, which is an autosomal recessive neurodegenerative disorder characterized by psychomotor regression and epilepsy with variable degree of liver involvement. Moreover, it is known that NARS2 mutations cause various clinical phenotypes, including non-syndromic hearing loss, Leigh syndrome, intellectual disability with epilepsy and severe myopathy. The individuals with PARS2 and NARS2 mutations, we have reported here demonstrate similar neurological features as those previously reported, with diversity in clinical presentation such as hearing loss and seizure type. Our data broaden the clinical and mutational spectrum of PARS2- and NARS2-related disorders.
Similar content being viewed by others

Introduction
The human genome contains 19 genes that encode aminoacyl-tRNA synthetases (aaRSs) for mitochondrial translation.1 Each aaRS charges a specific tRNA molecule with its cognate amino acid. This reaction is an essential and fundamental process for integrity of mitochondrial protein synthesis, including for subunits of the mitochondrial oxidative phosphorylation complex, which facilitate cellular energy production in the form of adenosine triphosphate.2 Oxidative phosphorylation dysfunction caused by mutations in the genes encoding aaRSs results in clinically and genetically heterogeneous mitochondrial diseases.3, 4 Recently, biallelic mutations in two mitochondrial aaRS genes, prolyl-tRNA synthetase 2 (PARS2; NM_152268.3) and asparaginyl-tRNA synthetase 2 (NARS2; NM_024678.5), were reported in individuals with variable clinical manifestations in one and four unrelated families, respectively.5, 6, 7 However, their pathogenicity in relation to clinical spectrum has not been validated. Here we performed whole-exome sequencing of four unrelated families with infantile-onset neurodegenerative phenotypes, and identified novel mutations of PARS2 in one family and NARS2 in the other three. These individuals are clinically evaluated and reviewed.
Materials and methods
Written informed consent was obtained from all participants. This study was approved by the Institutional Review Boards of Yokohama City University School of Medicine and Showa University School of Medicine.
Whole-exome sequencing
Whole-exome sequencing was performed in all index patients. DNA samples were captured by SureSelectXT Human All Exon V5 (Agilent Technologies, Santa Clara, CA, USA) and sequenced on the Illumina HiSeq2500 with 101 bp paired-end reads (Illumina, San Diego, CA, USA). Exome data processing, variant calling and annotation were performed as described previously.8 The averaged read-depth of protein-coding regions ranged from 86 to 128 ×, and at least 96% of target bases were sequenced by 10 or more reads for each patient. The variants that fulfilled the following criteria were considered for further analysis: (1) variants with minor allele frequency<1% in the Exome Sequencing Project (ESP6500), Exome Aggregation Consortium and in-house Japanese exome data sets; (2) possible pathogenicity based on mutation type (nonsense, missense, frameshift and splice site), with computational prediction of the deleterious protein function effect by SIFT, Polyphen-2, and MutationTaster; (3) biallelic mutations, at least one of that was deleterious under an autosomal recessive model; and (4) variants found in a gene whose mutations causes epileptic encephalopathy in the literature. Co-segregation of the candidate variants with disease in each family was examined by direct Sanger sequencing.
Each variant was validated by Sanger sequencing. In individuals 3 and 4, total RNA was extracted from Epstein–Barr virus–transformed lymphoblastoid cells, and used to synthetized complementary DNA. NARS2 complementary DNA was amplified using primers located at exons 4 and 6. Primer sequences are available on request.
Web resources
URLs of the web resources used are:
Exome Aggregation Consortium (ExAC) Browser, http://exac.broadinstitute.org/.
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/.
SIFT, http://sift.jcvi.org/.
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/.
MutationTaster, http://MutationTaster.org/.
Results
The clinical manifestations are described as follows and summarized in Table 1 and Supplementary Table S1.
Family 1
Individual 1, a 9-year-old girl, was delivered at 37 gestational weeks without asphyxia. She showed early developmental delay. She could control her head at 5 months of age and sit at 3 years, but was unable to walk unaided or speak any meaningful words by 9 years. At 5 months, she developed infantile spasms with hypsarrhythmia evident by electroencephalogram (EEG), supporting a diagnosis of West syndrome. She was treated effectively with adrenocorticotropic hormone injection, but an abnormal EEG persisted with occasional facial spasms (Supplementary Figure S1a). These were temporarily responsive to anti-epileptic drugs, but complex partial seizures were observed monthly at 9 years old. She had profound intellectual disability (developmental quotient 10) and hypotonia. At 6 years, she had a short stature (95.2 cm, −4.0 s.d.) and postnatal microcephaly (44.1 cm, −3.9 s.d.). At 7 years, brain magnetic resonance imaging (MRI) showed cerebral white matter hypomyelination and diffuse cortical atrophy with frontal lobe volume loss (Supplementary Figure S1c). Lactate elevation, up to 28.9 mg dl−1 in blood and 17.7 mg dl−1 in cerebrospinal fluid (normal range: 4.2–17.0 mg dl−1), was detected at 6 months of age.
Individual 2, a 3-year-old girl, is the younger sister of individual 1. She was born at 39 weeks of gestation without asphyxia. Her early development was normal until 4 months of age when she showed epileptic spasms. Left or right independent occipital-dominant multifocal or sometimes diffusely propagating sharp waves or sharp-and-slow waves were observed on EEG (Supplementary Figure S1b). Lactate levels in blood increased up to 27.4 mg dl−1 (normal range: 4.2–17.0 mg dl−1). She was diagnosed with infantile spasms without hypsarrhythmia. Adrenocorticotropic hormone therapy was effective on her spasms, but psychomotor regression was observed. Head control was lost in the prone position after seizure onset. She exhibited intellectual disability (developmental quotient 50) and postnatal microcephaly (38.0 cm, −2.4 s.d. at 4 months), along with decreased frontal lobe volume and delayed subcortical white matter myelination on brain MRI at 16 months of age (Supplementary Figure S1d). Hearing and visual impairments were not apparent in either sibling. Increased deep tendon reflexes (DTRs) of lower limb were observed, although upper reflexes were normal in either sibling. Babinski sign was negative.
Family 2
Individual 3, an 8-year-old boy, was delivered at term without any complications. His early development was slow and he achieved his maximum developmental level (with head control and rolling over) by 8 months. After an episode of acute bronchitis at 8 months, he showed poor weight growth and psychomotor regression. Hearing impairment was observed at 13 months. Further, he developed status epilepticus at 14 and 19 months. Lactate levels in blood and cerebrospinal fluid were in the normal range (12.6 and 10.6 mg dl−1, respectively; normal range: 5.0–20.0 mg dl−1). EEG demonstrated diffuse spikes and slow-wave complexes. Afterwards, he repeatedly suffered from respiratory infections and required oxygen inhalation or mechanical ventilation. Brain MRI was normal at 19 months of age, but computed tomography showed diffuse brain atrophy at 3 years. By 8 years old, his height was 100 cm (−9.5 s.d.), weight 9.5 kg (−3.2 s.d.) and occipitofrontal circumference 47 cm (−2.5 s.d.). He presented with severe intellectual disability and flaccid quadriplegia. An upper and lower limb DTRs were absent. Blood lactate levels were elevated (25.8 mg dl−1; normal range: 3.3–14.9 mg dl−1) and urine test showed proteinuria. Optic nerve atrophy was noted. Brain MRI at 8 years showed progression of diffuse atrophy (cortical, white matter, basal ganglia, thalamus and cerebellum) and marked T2 hyper-intensity signals in the white matter, basal ganglia and thalamus (Supplementary Figures S2a–c).
Individual 4, a 1-year-old girl, is the younger sister of individual 3. She was delivered at term without complications. She could control her head at 3 months of age and rolled over at 4 months. After an episode of respiratory infection at 10 months, she exhibited swallowing difficulties. She then showed developmental regression with muscle weakness and hypotonia. At 14 months of age, she had myoclonic seizures. At 15 months, her weight was 6.8 kg (−3.0 s.d.), height 74 cm (−1 s.d.) and occipitofrontal circumference 42 cm (−2.5 s.d.). She showed intellectual disability and flaccid quadriplegia. DTRs were absent in the extremities. Lactate was slightly increased, up to 22.8 mg dl−1 in blood and 16.5 mg dl−1 in cerebrospinal fluid (normal range: 3.3–14.9 mg dl−1). Auditory brainstem response test revealed severe bilateral hearing impairment, and EEG showed multifocal spikes. Brain MRI was normal.
Family 3
Individual 5, a 2-year-old girl, was born at 37 weeks of gestation after an uneventful perinatal period. Her psychomotor development was severely delayed. She could control her head at 4 months of age, but achieved no further developmental milestones. She developed hemi-convulsive status epilepticus in her right side at 8 and 11 months. She also developed daily absence seizures and frequent myoclonic seizures after 13 months. Brain MRI was normal. EEG showed frequent spikes and wave complexes in the left occipital area while awake, and modified hypsarrhythmia during sleep. Auditory brainstem response test revealed severe bilateral hearing impairment. Serum lactate was slightly increased (18.1 mg dl−1; normal range: 3.0–17.0 mg dl−1). She developed right-sided hemi-convulsive status epilepticus again at 18 months of age. Thereafter, hemiparesis remained in her right upper and lower limbs. Brain MRI revealed diffuse atrophic changes in the left cerebrum (Supplementary Figure S2d). These findings suggest hemiconvulsion–hemiplegia–epilepsy (HHE) syndrome, which is characterized by prolonged unilateral seizures followed by development of hemiplegia and focal epilepsy with cerebral atrophy of one hemisphere.9 She showed intellectual disability (developmental quotient 57) and gradually developed muscle weakness in her all extremities and pharynx. DTRs of the extremities were decreased.
Family 4
Individual 6, a 4-year-old boy, is the first child to parents of Druze origin from Israel. He was born at 38 weeks of gestation with no asphyxia. He presented with mild axial hypotonia at 4 months of age. He developed generalized tonic-clonic seizures, myoclonic seizures and status epilepticus at 5 months. EEG showed a burst suppression pattern. After the first seizures, he presented with psychomotor regression. At 9 months, brain MRI showed cerebral atrophy with extended vacuolization of the periventricular white matter, basal ganglia, corpus callosum and cerebellum (Supplementary Figures S2e–g). Auditory brainstem response test revealed severe bilateral hearing impairment. At 12 months, he presented with severe hypertonia and scissor legs. Lactate in blood, but not cerebrospinal fluid, was elevated at 33 mg dl−1 (normal range: 2–20 mg dl−1). He presented with normal DTRs at the age of 6 months, but the hyperreflexia including babinski was observed at age 2.5years. At 3 years of age, his weight was 9.5 kg (−3.2 s.d.) and occipitofrontal circumference 47 cm (−2.5 s.d.). He had intellectual disability and spastic quadriplegia.
Genetic analyses
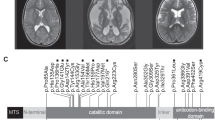
We identified two and five novel variants in PARS2 and NARS2, respectively. Six (out of the seven) were missense variants: c.283G>A (p.Val95Ile) and c.607G>A (p.Glu203Lys) in PARS2, and c.151C>T (p.Arg51Cys), c.500A>G (p.His167Arg), c.707T>G (p.Phe236Cys) and c.1184T>G (p.Leu395Arg) in NARS2. One canonical splice site variant in NARS2 was also found (Figures 1a and b). Computational programs supported deleterious effects of the missense variants (Supplementary Table S2). Reverse transcription PCR was performed to evaluate the NARS2 splice site variant, c.594+1G>A. An abnormal short PCR product was obtained from lymphoblastoid cells of individuals 3 and 4 only, and not from a normal control. Sanger sequencing of this abnormal PCR fragment revealed an 81- bp deletion lacking the entire exon 5, In-frame 27 amino-acid deletion (p.Asp172_Glu198del) would be expected (Figure 1c).

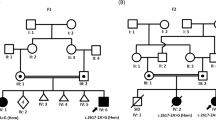
PARS2 and NARS2 mutations identified in this study. (a) Pedigree and PARS2 mutations in family 1. Mutations are positioned alongside protein functional domains. (b) Pedigrees and NARS2 mutations in families 2–4. Mutations are positioned alongside protein functional domains. (c) RT–PCR products (involving exons 4–6) using lymphoblastoid cell complementary DNA from individuals 3 and 4, and a healthy control. Abnormal RT–PCR products were observed in patients, suggesting that c.594+1G>A affects splicing. Sequencing of the aberrant RT–PCR product shows that this mutation causes skipping of exon 5. Topmost band in patients correspond to heteroduplex dsDNA comprising of wild-type and mutant allele (c.594+1G>A), which was cleaved by structure-selective T7 endonuclease I (data is not show). dsDNA, double-stranded DNA; RT–PCR, reverse transcription PCR.
All these variants are appropriately inherited on the basis of an autosomal recessive model (Figures 1a and b).
Discussion
All seven variants identified in this study were novel. Not all of missense variants were deleterious by three programs (Supplementary Table S2). The p.Val95Ile mutation of PARS2 and p.Phe236Cys mutation of NARS2 were predicted to be deleterious by one (MutationTaster) and two (SIFT and MutationTaster) computational tools, respectively. Nonetheless, aaRSs are essential for cell function and development in humans; therefore, even mildly impaired function from recessive mutations can cause mitochondrial disease.4 The p.Arg51Cys mutation is located within the anticodon-binding domain, although the others are located within the core catalytic domain, suggesting that variants in both domains have the same direction of functional effects associated with similar neurodegenerative disorders. The variants identified in this study were absent from, or extremely rare, in Exome Aggregation Consortium (Supplementary Table S2): c.283G>A of PARS2 was present at the highest frequency in East Asian population (0.161% compared with 0.0132% in total).
The main clinical features of our patients include epilepsy, variable degree of developmental delay and progressive developmental regression followed by hypotonia, postnatal microcephaly and intellectual disability. Diffuse cerebral atrophy was typically seen (except for individual 4) at a variable degree, as well as atrophy of the basal ganglia and cerebellum. Individual 4 (currently 1 year old) might develop cortical atrophy in later stages, as did her affected brother. There are several distinctive clinical features. Individuals with NARS2 but not PARS2 mutations consistently had sensorineural hearing impairments. Infantile spasms and myoclonic seizures were associated with PARS2 and NARS2 mutations, respectively. Interestingly, individual 5 developed HHE syndrome. Although the etiology of HHE remains largely unknown, the role of some genetic factors have been suggested.10 Our data imply that NARS2 defects might be a risk factor for HHE syndrome.
Associated cardiomyopathy and renal impairments were reported in Alpers’ syndrome with PARS2 and NARS2 mutations, respectively.6, 11 Neither circulatory collapse nor severe renal dysfunction was recorded in our patients. Macrosomia (+5 s.d. for height) was described previously as a unique feature in an individual with PARS2 mutations.6 Our individual with PARS2 mutations did not show macrosomia.
In conclusion, a broad range of clinical features is associated with PARS2 and NARS2 mutations. More patients are needed to draw clearer phenotype–genotype correlations.
Change history
16 February 2017
This article has been corrected since Advance Online Publication, and an erratum is also printed in this issue.
References
Suzuki, T., Nagao, A. & Suzuki, T. Human mitochondrial tRNAs: biogenesis, function, structural aspects, and diseases. Annu. Rev. Genet. 45, 299–329 (2011).
Ylikallio, E. & Suomalainen, A. Mechanisms of mitochondrial diseases. Ann. Med. 44, 41–59 (2012).
Antonellis, A. & Green, E. D. The role of aminoacyl-tRNA synthetases in genetic diseases. Ann. Rev. Genomics Hum. Genet. 9, 87–107 (2008).
Konovalova, S. & Tyynismaa, H. Mitochondrial aminoacyl-tRNA synthetases in human disease. Mol. Genet. Metab. 108, 206–211 (2013).
Simon, M., Richard, E. M., Wang, X., Shahzad, M., Huang, V. H., Qaiser, T. A. et al. Mutations of human NARS2, encoding the mitochondrial asparaginyl-tRNA synthetase, cause nonsyndromic deafness and Leigh syndrome. PLoS Genet. 11, e1005097 (2015).
Sofou, K., Kollberg, G., Holmstrom, M., Davila, M., Darin, N., Gustafsson, C. M. et al. Whole exome sequencing reveals mutations in NARS2 and PARS2, encoding the mitochondrial asparaginyl-tRNA synthetase and prolyl-tRNA synthetase, in patients with Alpers syndrome. Mol. Genet. Genomic Med. 3, 59–68 (2015).
Vanlander, A. V., Menten, B., Smet, J., De Meirleir, L., Sante, T., De Paepe, B. et al. Two siblings with homozygous pathogenic splice-site variant in mitochondrial asparaginyl-tRNA synthetase (NARS2. Hum. Mut. 36, 222–231 (2015).
Saitsu, H., Nishimura, T., Muramatsu, K., Kodera, H., Kumada, S., Sugai, K. et al. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat. Genet. 45, 445–449 (2013).
Auvin, S., Bellavoine, V., Merdariu, D., Delanoe, C., Elmaleh-Berges, M., Gressens, P. et al. Hemiconvulsion-hemiplegia-epilepsy syndrome: current understandings. Eur. J. Paediatr. Neurol. 16, 413–421 (2012).
Yamazaki, S., Ikeno, K., Abe, T., Tohyama, J. & Adachi, Y. Hemiconvulsion-hemiplegia-epilepsy syndrome associated with CACNA1A S218L mutation. Pediatr. Neurol. 45, 193–196 (2011).
Sofou, K., Moslemi, A. R., Kollberg, G., Bjarnadottir, I., Oldfors, A., Nennesmo, I. et al. Phenotypic and genotypic variability in Alpers syndrome. Eur. J. Paediatr. Neurol. 16, 379–389 (2012).
Acknowledgements
We thank all the individuals and their families for participating in this study. We also thank Ms N Watanabe, M Sato and K Takabe for their technical assistance. This work was supported by grants from Research on Measures for Intractable Diseases; Comprehensive Research on Disability Health and Welfare; the Strategic Research Program for Brain Science, Practical Research Project for Rare/Intractable Diseases, the Initiative on Rare and Undiagnosed Diseases in Pediatrics and the Initiative on Rare and Undiagnosed Diseases in Adults from the Japan Agency for Medical Research and Development; a grant-in-aid for Scientific Research on Innovative Areas (Transcription Cycle) from the Ministry of Education, Culture, Sports, Science and Technology of Japan; grants-in-aid for Scientific B and C from the Japan Society for the Promotion of Science; and the fund for Creation of Innovation Centers for Advanced Interdisciplinary Research Areas Program in the Project for Developing Innovation Systems from the Japan Science and Technology Agency; and the Takeda Science Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Rights and permissions
About this article

Cite this article
Mizuguchi, T., Nakashima, M., Kato, M. et al. PARS2 and NARS2 mutations in infantile-onset neurodegenerative disorder. J Hum Genet 62, 525–529 (2017). https://doi.org/10.1038/jhg.2016.163
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2016.163
This article is cited by
-
Novel NARS2 variants in a patient with early-onset status epilepticus: case study and literature review
BMC Pediatrics (2024)
-
Novel NARS2 variant causing leigh syndrome with normal lactate levels
Human Genome Variation (2022)
-
Novel phenotype and genotype spectrum of NARS2 and literature review of previous mutations
Irish Journal of Medical Science (1971 -) (2022)
-
Four pedigrees with aminoacyl-tRNA synthetase abnormalities
Neurological Sciences (2022)
-
Novel variants in the NARS2 gene as a cause of infantile-onset severe epilepsy leading to fatal refractory status epilepticus: case study and literature review
neurogenetics (2021)

{kind=link}
{kind=link}