
Abstract
Similar to plant growth, soil carbon (C) cycling is constrained by the availability of nitrogen (N) and phosphorus (P). We hypothesized that stoichiometric control over soil microbial C cycling may be shaped by functional guilds with distinct nutrient substrate preferences. Across a series of rice fields spanning 5–25% soil C (N:P from 1:12 to 1:70), C turnover was best correlated with P availability and increased with experimental N addition only in lower C (mineral) soils with N:P⩽16. Microbial community membership also varied with soil stoichiometry but not with N addition. Shotgun metagenome data revealed changes in community functions with increasing C turnover, including a shift from aromatic C to carbohydrate utilization accompanied by lower N uptake and P scavenging. Similar patterns of C, N and P acquisition, along with higher ribosomal RNA operon copy numbers, distinguished that microbial taxa positively correlated with C turnover. Considering such tradeoffs in genomic resource allocation patterns among taxa strengthened correlations between microbial community composition and C cycling, suggesting simplified guilds amenable to ecosystem modeling. Our results suggest that patterns of soil C turnover may reflect community-dependent metabolic shifts driven by resource allocation strategies, analogous to growth rate–stoichiometry coupling in animal and plant communities.
Similar content being viewed by others

Introduction
Although microbial communities are critical to carbon (C) flow in the biosphere, ecosystem models have only recently begun to simulate variation in their metabolism in soils (Li et al., 2014; Wang et al., 2015; Weider et al., 2015). Increasingly powerful and available data on microbial community structure and function might help to better inform these efforts (McGuire and Treseder, 2010; Singh et al., 2010; Schimel and Schaeffer, 2012; Graham et al., 2016). Yet, compared with taxonomically constrained processes such as ammonia or methane oxidation (Bouskill et al., 2012; Ho et al., 2013), delineation of functional groups for decomposition in soils is challenging due to the broad distribution and functional redundancy of the relevant traits (Allison and Martiny, 2008; Schimel and Schaeffer, 2012; Berlemont and Martiny, 2013; Martiny et al., 2015). However, consideration of interactions between C and nutrient cycling by soil microbes may help clarify ecologically relevant functional guilds. For example, consistent shifts in terrestrial soil C turnover and retention with mineral N addition (Craine et al., 2007; Treseder, 2008) appear mediated in part by increased abundance of microbes utilizing simple C substrates, at the expense of those mining complex aromatic C and organic N (Treseder et al., 2011; Fierer et al., 2012; Ramirez et al., 2012; Cederlund et al., 2014; Amend et al., 2015).
Phosphorus (P) availability can also contribute to regulation of soil C cycling, and the underlying traits for microbial P cycling could analogously be linked with traits for C and N acquisition. Although less commonly studied than soil C and N interactions, P availability may affect land C sink strength at ecosystem and global scales (Wang et al., 2007; Vitousek et al., 2010; Goll et al., 2012) and contribute to the regulation of soil C turnover rates even in nominally N-limited habitats, such as grasslands, temperate forests and leaf litter (Bradford et al., 2008; Manzoni et al., 2010; Strickland et al., 2010; Fisk et al., 2015). Soil P availability may not only affect the biomass of soil microbes (Griffiths et al., 2012; Zhang et al., 2015) but critically might also control community-scale rates of metabolism (Strickland et al., 2010; Spohn and Chodak, 2015), which could reflect underlying stoichiometric constraints at the scale of individual cells (Hartman and Richardson, 2013).
As posited by the growth rate hypothesis (Elser et al., 1996, 2000, 2003), cellular growth rates are linked to biomass N:P ratios by the high P demands of ribosomal RNA, which determines in part the rate of synthesis of N-rich proteins. Differentiation of organism C, N and P demands based on growth rate variation forms the foundation of Ecological Stoichiometry theory (Sterner and Elser, 2002; Vrede et al., 2004; Allen and Gillooly, 2009), enabling community shifts under different nutrient regimes to be connected with predictable alterations in ecosystem C cycling, particularly in aquatic ecosystems (Sterner and Elser, 2002; Weber and Deutsch, 2010; Follows and Dutkiewicz, 2011; Hessen et al., 2013; Mock et al., 2015). Development of a parallel framework to link stoichiometric regulation of microbial metabolism to soil C cycling at the community and ecosystem scales is highly desirable (Hall et al., 2011; Sistla and Schimel, 2012; Zechmeister-Boltenstern et al., 2015), especially given the integration of stoichiometric regulation of primary producers and decomposition into current terrestrial ecosystem models (Yang et al., 2014; Reed et al., 2015).
However, the inter-relationships between soil stoichiometry, microbial communities and soil C cycling are not currently well understood. In culture, microbial growth rates are linked with cell N:P stoichiometry and ribosomal RNA content or gene copy number (Makino and Cotner, 2004; Karpinets et al., 2006; Keiblinger et al., 2010; Vieira-Silva and Rocha, 2010; Franklin et al., 2011), which can vary among bacterial lineages (Mouginot et al., 2014; Roller et al., 2016). Separately, variation or manipulation of P availability and stoichiometry in soils has been associated with shifts in microbial community composition (Güsewell and Gessner, 2009; Fanin et al., 2013; Leff et al., 2015; Spohn et al., 2015), without considering relationships to soil C cycling or metabolic differences among responsive microbes.
We postulated that ecosystem-scale relationships between soil C cycling and P availability may arise due to changes in microbial metabolism, which reflect a dependence of growth rates on P availability, and are underpinned by shifts in microbial communities based on differences in their nutrient utilization potential. We combined plot-scale manipulations and measurements of C cycling across a broad soil C:N:P gradient with high-throughput sequencing techniques to address specific hypotheses at different scales of ecological organization, including: (1) Soil C cycling rates are associated with P availability and stoichiometry at the ecosystem scale; (2) At the community scale, taxonomy and function of microbes are associated with soil P stoichiometry; (3) At the individual scale, genomic features and potential utilization of C, N and P substrates differ characteristically among taxa, and (4) Across these scales, rates of soil C cycling are linked with the abundance of microbial groups defined by differences in stoichiometric resource allocation. To enable evaluation of the latter two hypotheses and interrogate relationships across scales, we inferred the stoichiometric utilization potential of individual microbes in situ, using 16S rRNA and shotgun metagenome sequencing in combination with inference from existing reference genomes (Langille et al., 2013).
Materials and methods
Experimental design and measurement of soil C cycling and chemistry
Soils and C flux data were obtained from a ca. 120 hectare area under continuous rice cultivation since 2008 on Twitchell Island (38.1053 N, 121.6542 W) in the Sacramento-San Joaquin Delta, California, USA. Study plots (4 × 5 m2) were established to test N fertilization effects across a gradient of soil C content in Rindge mucky silt loam soils (Typic Halposaprists). Additional details of this study and site, including crop yield and plant N uptake, are described by Espe et al. (2015). Fertilized plots had solid urea pellets broadcast at a single time point within 5 days of flooding on 15 May 2013.
A subset of these plots was used to test effects of added N on trace greenhouse gas fluxes in a complete randomized block design with four replicate plots each for control (0 N) and 80 kg N ha−1 fertilization (80 N) treatments in each of four fields with ca. 5, 10, 20 and 25% soil C content (32 total plots). Fluxes of CO2, CH4 and N2O in closed vented soil chambers were measured weekly for the calendar year 2013, with complete methods and data described in a companion study (Ye et al., 2016).
For each plot across the soil C gradient, CO2 fluxes were integrated over the inundated period of the rice-growing season (15 May–10 August; Supplementary Figure S1) to match soil collection date, instead of using closely correlated annual fluxes (Ye et al., 2016). We then calculated soil C turnover, the proportion of soil C content (in the top 15 cm of soil) respired over the growing season as CO2 (Σ g CO2 m−2 per g soil C m−2) to account for differences in bulk C supply on soil respiration.
Soil samples were obtained from each of the experimental plots between rows of rice at the end of the growing season (9–10 August 2013), just prior to water drawdown and timed to coincide with peak flooded soil CO2 and CH4 emissions (Supplementary Figure S1, Ye et al., 2016). Three replicate topsoil samples were taken from each plot 0–15 cm beneath the soil surface, corresponding approximately to the soil horizon (anthraquic horizon) affected by agricultural activity in paddy soils (Kögel-Knabner et al., 2010), which also harbors the most abundant populations of bacteria (Lee et al., 2015).
Soils from each plot were composited and analyzed for pH, chloride and sulfate concentrations, total pools of C, N and P and extractable NH4+, NO3− and PO42−. Further details on soil collection, processing, biogeochemistry and greenhouse gas fluxes are provided in Supplementary Methods and Supplementary Table S1. The relative abundances of C compound classes were determined in lyophilized soils using Fourier-transformed infrared spectroscopy (attenuated total reflection Fourier-transformed infrared spectroscopy), with 64 scans by a Thermo-Scientific (Waltham, MA, USA), Nicolet iS50 FT-IR and peak assignments were made following Parikh et al. (2014).
Microbial community sequencing and data processing
Soil DNA was extracted from 0.5 g of homogenized composite sample from each plot using a MoBio Laboratories (Carlsbad, CA, USA) PowerLyzer PowerSoil DNA Extraction Kit. For metagenomic shotgun sequencing, ~300 bp insert indexed libraries (Kapa Biosystems, Wilmington, MA, USA) were pooled 12 per lane on an Illumina (San Diego, CA, USA) HiSeq 2500 using TruSeq SBS v4 chemistry to produce 150 bp length paired-end reads, yielding ~5.0 Gbp per sample after contaminant and quality filtering (Supplementary Methods, Supplementary Table S2). Sequences were annotated via the Integrated Microbial Genomes (IMG) pipeline (Huntemann et al., 2015) (details in Supplementary Methods). Gene abundance data calculated using the clusters of orthologous group (COG) ontology (Galperin et al., 2015) and adjusted for contig depth were downloaded from JGI’s IMG database (Markowitz et al., 2014) as a gene count by sample table from samples stored under IMG Genome IDs 3300005873–3300005905. Across our samples, ca. 42% of genes were assigned to COGs (Supplementary Table S2); others were not considered further.
Reads aligning to large subunit ribosomal RNA genes of bacterial, archaeal, fungal and other eukaryotic origins were identified and classified using the METAXA2 pipeline (Bengtsson-Palme et al., 2015).
Bacterial and archaeal community composition was determined from soil DNA extracts by amplifying the V4 region of 16S rRNA using barcoded multiplexed primers 515F and 816R (Caporaso et al., 2012; Tremblay et al., 2015). Sequences were processed using a standard pipeline (Tremblay et al., 2015) yielding ca. 24 000 unique operational taxonomic units (OTUs) at depth of >74 000 reads per sample (details in Supplementary Methods).
For imputation of community function by PICRUSt (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States) (Langille et al., 2013), 16S rRNA sequences were mapped at 97% sequence identity to the GreenGenes reference database (DeSantis et al., 2006) for which PICRUSt provides precomputed functional profiles, matching ca. 46% of sample reads and 48% of abundant OTUs (>250 total reads) (Supplementary Table S2). Abundance data from 16S sequences were corrected for ribosome copy number bias before predicting the abundance of COG ontology functions in soil metagenomes using PICRUSt. Imputed and sequenced metagenome COG function tables were rarified separately and then both normalized to 10 million COGs/sample to compare accuracy of PICRUSt predictions using the evaluate_test_datsets.py function.
Statistical data analysis
Statistical relationships were evaluated using packages in the R statistical program (R Core Team, 2015) and Quantitative Insights Into Microbial Ecology (QIIME) software (Caporaso et al., 2010). Relationships between log-transformed soil biogeochemical data, soil CO2 fluxes and C turnover were screened using linear regression, and selected relationships were evaluated using standardized major axis regression, with tests of N fertilization interactions (R: SMATR 3 package). Soil C chemistry (by Fourier-transformed infrared spectroscopy) was compared with nutrients and C turnover using partial least squares regression (R: pls package).
Relationships between C turnover and the abundance of metagenome functions and taxonomic categories (aggregated at each taxonomic rank) were evaluated using Pearson’s correlations with control of false discovery rates (FDRs) at ⩽5% false positive (FDR⩽0.05). Shifts in the abundances of these functions were shown in heatmaps (R:Heatmap2) using log-transformed COG count data, which were standardized (z-score scaled) across all soil plots. Aggregated shifts in C, N and P metabolism (categorical hierarchy in Supplementary Table S3) were compared with soil N:P ratios using log, Box–Cox or polynomial transformations prior to standardized major axis regression. Phylogenetic patterns were visualized using the Interactive Tree of Life web interface (Letunic and Bork, 2016).
Abundance weighted UniFrac distances (Hamady et al., 2009) were used to evaluate relationships between phylogenetic community structure, soil chemistry and C turnover using Mantel’s tests, while N fertilization effects on community UniFrac distance were tested by analysis of similarities, each with 999 permutations of samples (R: vegan). Fertilization responses of log-transformed OTU and taxonomic group abundances were tested with indicator species analysis (R: vegan) and using analysis of variance models with FDR⩽0.05 (QIIME: category_significance.py).
Trait differentiation and gradient responses of imputed function
Microbial genera correlated with soil C turnover (FDR<0.05) were recoded as either positively or negatively correlated, and shrinkage linear discriminant analysis (R: sda) was used to test differences among these groups in PICRUSt-imputed pathways of C, N and P substrate metabolism (Supplementary Table S3) and ribosome copy numbers.
We calculated the relative ‘allocation’ of genes for utilizing available or recalcitrant C, N and P substrates for each OTU in our soils, based on ratios of metabolic categories (Supplementary Table S3). Each OTU was scored by its ratio of imputed copy numbers of carbohydrate to aromatic and carboxylic utilization functions (Ccarb:arom), while nitrogen utilization was scored on assimilatory vs dissimilatory processes (Ndiss:assim) following Helton et al. (2015), and P functions were scored based on ratios of scavenging (for example, phosphatases) vs uptake (for example, P transporters) genes (Pscav:uptk). Details of functional genes and categories can be found in Supplementary Methods and Supplementary Table S3. Patterns of C, N and P substrate allocation for each OTU were plotted on the three axes Ccarb:arom, Ndiss:assim and Pscav:uptk with confidence intervals for major taxonomic groups (R: plot3Drgl). ADONIS non-parametric modeling (R: vegan) was used to test the sequential contribution of taxonomic ranks to the three-dimensional C, N and P allocation of microbial OTUs, using standardized and centered scores.
Relationships between these functional traits (including ribosome copy number) and microbial community composition were evaluated using a linear model for community assembly by trait selection (CATS) with the mvabund R package (Warton et al., 2015). The CATS model represents taxon count data as a function of environmental and trait variables, with significant trait by environment interactions (filtered by LASSO) indicating ‘trait selection’.
De novo functional ‘guilds’ were formed using K-means clustering (R: vegan) of preselected functional genes related to C, N and P substrate metabolism (Supplementary Table S3). Comparisons of microbial community structure (16S UniFrac distance) and de novo functional ‘guilds’ as predictors of trends in soil C cycling were made using Mantel’s tests to determine correlations between differences in community or guild structure and C turnover using Bray–Curtis dissimilarity matrices. Guild abundances were also weighted (multiplied) by their mean ribosome copy number and/or Ccarb:arom allocation values prior to additional Mantel’s tests to evaluate how accounting for these traits might improve relationships between guild abundances and soil C turnover.
Results
Soil C cycling mediated by N:P availability and C quality
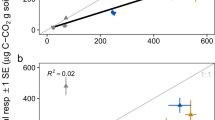
Soil respiration varied significantly along the soil C gradient, with effects of N fertilization appearing contingent on ambient nutrient availability. The highest cumulative CO2 fluxes occurred at intermediate levels of soil C (Figure 1a, Supplementary Figure S1), while soil C turnover (Σ g CO2−C m−2 per g soil C m−2) decreased with soil C content and total N:P ratios (Figures 1b and d), consistent with global-scale patterns of higher N:P ratios with increasing soil C content (Figure 1c). Soil C turnover was also positively correlated with extractable inorganic P (Figure 1e,Table 1). N fertilization increased both CO2 production and C turnover only in lower C soils (5% and 10% C; here termed ‘mineral’), where N:P ratios were less than the ratio of 16:1 by Redfield (1958) (Figures 1a, b and d). Soil C compounds also varied with C content along the soil gradient, with mineral soils containing relatively more carbohydrates and fewer aromatic and carboxylic compounds (Figure 1f, Supplementary Figure S2) than higher C soils (20 and 25% C; here termed ‘organic’).

C, N and P interactions shape soil CO2 flux and soil C turnover. Effects of soil C content and added N on cumulative growing season CO2 fluxes (a) and C turnover (b) from rice soils. (c) Soil N:P ratios as a function of soil C in rice study plots (colors) and globally (gray). Soil C turnover as a function of (d) total N:P ratios and (e) soil available (Olsen) P. Correlation coefficients (R2) and dashed lines in (b, d and e) show fit of ANCOVA models (x+N treatment) using Standardized Major Axis (Type II) regression. (f) Fourier-transformed infrared spectra of soil organic matter, framed by high (dried leaf tissue) and low (clay) C reference materials, colored by field% C as in other panels. Spectra from control soils are shown as solid lines while N-fertilized treatments are shown as dashed lines. Asterisks in (a, b, d and e) show differences (analysis of variance, P⩽0.05) among control (0 N) and 80 kg N ha−1 treatments (80 N) tested separately for each rice field.
Shifts in C, N and P substrate metabolism associated with C turnover
Major classes of C, N and P elemental metabolism in shotgun metagenome data varied with N:P ratios along the soil C gradient (Figure 2a, Table 2), with steeper shifts in the relative abundances of aggregated functions for C, N and P metabolism appearing near the Redfield ratio (N:P=16:1). With increasing soil N:P ratios (and soil C content), dominant metabolic pathways shifted from carbohydrate to aromatic/carboxylic metabolism and from N dissimilation to assimilation, while P scavenging potential increased (phosphatases, phytases and phosphonate utilization; Figure 2a, Supplementary Figures S3–5). Conversion of these data into ratios representing preference for more or less readily available substrates (see Materials and methods section) revealed increasing use of more resistant C, N and P forms with increasing N:P along the soil C gradient (Supplementary Figures S6a–c, Supplementary Table S4). C, N and P substrate utilization patterns were also intertwined, with carbohydrate degradation linked to denitrification and P transport and aromatic/carboxylic utilization linked with N assimilation and P scavenging (Supplementary Figures S6d–f, Supplementary Table S5).

Microbial C, N and P substrate use linked with stoichiometry and soil C turnover. Metabolic classes share color schemes in both panels, which compare their abundances in shotgun sequence and PICRUSt imputed data for each study plot. (a) Abundance of aggregated metabolic functional classes for C (top), N (middle) and P cycling varied strongly with soil N:P ratios. Vertical scales (aggregate ortholog counts) are displayed for each metabolic category, with additional scales for PICRUSt-imputed data given in small italics where needed to align data trends but absent if data were on the same scale as metagenome sequence counts. (b) Relative abundance of specific metabolic functions correlated with soil C turnover (Pearson’s r⩾0.7; FDR<0.05) among rice soils, with both positively and negatively correlated functions shown. Additional functions for C, N and P metabolism that were not correlated with soil C are shown in Supplementary Figures S3–S5.
Higher rates of soil C turnover in mineral soils were associated with more abundant carbohydrate degradation genes and inversely correlated with the abundance of aromatic compound utilization and small carboxylic (TRAP) transporter genes (Figure 2b, Supplementary Figure S3). Soil C turnover was also correlated with the metagenomic abundance of dissimilatory nitrate reduction and negatively correlated with P uptake and P scavenging (alkaline and acid phosphatases, phytase) genes (Figure 2b, Supplementary Figures S3–S5).
Metabolic functions predicted based on 16S profiles (using PICRUSt) were largely consistent (Spearman ρ2=0.82) with shotgun metagenome annotations at the plot scale (Supplementary Table S6), and similarly predicted higher C turnover was linked with more genes for carbohydrate utilization but fewer for carboxylic/aromatic utilization or P scavenging (Figure 2a), though not all patterns were reproduced, particularly for N metabolism.
Total eukaryotic and fungal abundances based on large subunit rRNA sequences in our metagenome data were generally low (0.9–3.9 and 0.1–1.4%, respectively; Supplementary Table S2).
Microbial communities varied with C content and turnover, not N addition
Rice soil microbial community membership at the end of the growing season was not significantly altered by N fertilization at any level of soil C (Supplementary Figures S7a and d), from the perspective of community structure (UniFrac distances), taxonomic groups or individual OTUs. Changes in microbial community structure were correlated with each chemical factor along the soil C gradient but were most closely aligned with soil C, pH and P stoichiometry (Table 1), including soil C:P, N:P and extractable inorganic N:P. Partial Mantel’s tests excluding variance attributable to main study factors of soil C and (co-varying) pH showed significant, independent relationships between microbial community structure and soil stoichiometry, total P and inorganic P (Table 1) but not total or inorganic N forms. Soil C turnover was moderately correlated with shifts in microbial community structure (UniFrac distance; Mantel’s r=0.42), with residual variance attributable to soil stoichiometry and inorganic P (Table 1).
The abundances of several lineages were correlated with soil C turnover (Figure 3a, Supplementary Figure S7b and c, Supplementary Table S7) including Actinobacteria (primarily genera within the order Actinomycetales), Chloroflexi (Anaerolineae and Ktedonobacteria) and Fibrobacteres. The phyla Actinobacteria, Chloroflexi and Firmicutes also had significantly more OTUs that were positively correlated with C turnover than were negatively correlated (Figure 3a, Supplementary Figures S7b and c). Phyla that were negatively correlated with soil C turnover included Euryarchaeota, Gemmatimonadetes and Planctomycetes (except Isosphaeraceae).

Imputed C, N and P substrate use and genomic features vary with phylogeny and among OTUs correlated with soil C turnover. (a) Phylogenetic patterns of soil microbes associated with soil C turnover (shown at OTU level and higher taxonomic ranks, R2⩾0.5, P⩾0.05) with imputed genome features and potential use of different C, N and P substrates. Separate color legends are used for microbial taxa, their relationships with C turnover and functional categories of C, N and P metabolism; functional categories are colored as in Figure 1. (b) Imputed ribosome copy numbers were enriched among OTUs that were more strongly correlated with C turnover. (c) Functions enriched in OTUs correlated with higher (and lower) C turnover based on linear discriminant analysis. Bars showing significantly different traits are colored to match functional categories in Figure 1 and (a).
Imputed microbial traits associated with soil C turnover
OTUs with increasingly positive correlations to soil C turnover tended to have higher imputed ribosome copy numbers (Figure 3b), and linear discriminant analysis revealed that ribosome copies were higher on average in organisms positively correlated with C turnover (Figure 3c). In parallel to community-scale functional patterns (Figure 2), microbes that were positively correlated with C turnover had more abundant carbohydrate utilization and dissimilatory nitrate reduction genes (Figure 3c). High C turnover organisms also had less P scavenging and more abundant low affinity (pit) rather than high affinity (pst) phosphate transporter genes (Figure 3c).
C, N and P substrate utilization linked to community composition and C turnover
Comparison of imputed genome allocation to C, N and P substrates (Ccarb:arom, Ndiss:assim and Pscav:uptk) showed gross differences in substrate and element use strategies among OTUs (Figures 4a and b, Supplementary Figure S8), with much of the variation attributable to differences among phylum (R2=0.46) and class (R2=0.79) level taxonomic groupings (Table 3).

Taxonomic differentiation in C, N and P allocation contributes to microbial community assembly. (a) Taxonomic differences in stoichiometric allocation, where log-transformed axes for allocation among C, N and P substrates are derived from ratios of broad element classes and data points are individual PICRUSt-imputed genomes scaled by total COG count and colored by taxonomic groups. (b) 95% confidence intervals for the major taxonomic groups plotted as three-dimensional ellipsoids. Gray lines in (a and b) show (log) zero intercepts for each axis, representing 1:1 ratios for each allocation axis. Relationships between traits and taxa abundance in our ‘Community assembly via trait selection’ model (Warton et al., 2015) illustrated in (c) were used to evaluate significant environment × trait interactions in a joint species distribution model. (d) CATS model interaction terms between stoichiometric traits and soil chemical variables for rice soils, where color indicates the sign and intensity of interactions for traits aggregated at the order level.
Microbial traits for C, N and P allocation and ribosome copy number each had significant interactions with soil chemical variables in CATS ‘trait selection’ models (Warton et al., 2015) of the joint abundance of microbial orders across plots (Figures 4c and d). At each taxonomic rank, ribosome copy numbers were positively associated with inorganic P concentrations (Figure 4d, Supplementary Figure S9) and had weak negative association with nitrate. Soil nitrate concentrations had positive interactions with relative allocation to carbohydrates (Ccarb:arom), N dissimilation (Ndiss:assim) and, to a lesser extent, P scavenging (Pscav:uptk). Accounting for variation in these traits improved CATS community assembly models considerably compared with null models using only chemical predictors (Supplementary Table S8).
De novo functional groups based on clustering of similar patterns of potential C and nutrient use alone were not more predictive of differences in soil C cycling rates than were differences in 16S profiles (Table 4). However, weighting the abundance of these groups by simple growth rate-related traits, including ribosome copy number and carbohydrate allocation (Ccarb:arom), considerably improved correlations between soil C turnover and microbial ‘guilds’ (Table 4), particularly where N was added to soils (R2=0.75).
Discussion
Stoichiometric nutrient limitation of soil C cycling and metabolism
In the wetland soils studied, C cycling rates appeared dependent on the supply of soil C, N and P, yet constraints imposed by P availability featured most prominently. Non-linear patterns of soil CO2 fluxes reflected declining soil C turnover rates with increasing organic matter (Figures 1a and b). One explanation for this decline in soil C turnover is the lower availability of P relative to N as soil C accumulates (Figure 1c), consistent with global-scale patterns (Hartman and Richardson, 2013).
In agreement with our first hypothesis, soil C turnover was most closely associated with P availability, including N:P ratios and inorganic P (Table 1,Figures 1d and e). Moreover, we observed a breakpoint in N fertilization effects roughly corresponding to the Redfield ratio of N:P=16:1 (Figure 1d), which delineates a shift from N to P limitation (Redfield, 1958; Sterner and Elser, 2002; Cleveland and Liptzin, 2007). Similar to interacting effects of N and P additions on soil C cycling in upland soils (Nottingham et al., 2015; Poeplau et al., 2016), our results suggest N fertilization altered C cycling only where N was limiting relative to P. Importantly, these results are opposite of those expected from C:N ratios alone, which would predict N fertilization effects only in high C:N organic soils.
Alternately, declining C turnover at higher soil C may reflect accumulation of aromatic compounds, which are both resistant to degradation and hypothesized to directly inhibit decomposition (Melillo et al., 1982; Freeman et al., 2001). Accordingly, we found aromatic and carboxylic compounds were more abundant in higher C organic soils (Figure 1f). These shifts were mirrored by changes in microbial C substrate utilization potential in our metagenome data, which revealed more carbohydrate-degrading genes in mineral soils and greater potential utilization of complex C forms in organic soils (Figure 2, Supplementary Figure S6). Notably, aromatic compounds were most closely associated with increasing C:N, C:P and N:P ratios, opposite of inorganic P availability, while increasing soil C content was linked with greater abundance of both carbohydrates and aromatics (Supplementary Figure S2).
Despite its effects on C turnover, N fertilization did not significantly alter microbial metabolic potential or community structure in our rice soils (Figure 2, Supplementary Figure S7a and d), in contrast to upland soils where N fertilization consistently reduces soil C turnover and alters microbial communities (Ramirez et al., 2010; Fierer et al., 2011; Ramirez et al., 2012). Both nitrous oxide fluxes (Ye et al., 2016) and gene abundances for N dissimilation were higher in our mineral soils (Figure 2), which could suggest excess N availability consistent with their lower C:N ratios. However, N addition increased C turnover only in mineral soils (Figure 1) and did not alter in situ N2O fluxes in any of our soils (Ye et al., 2016). This decoupling of denitrification from N supply could indicate that denitrifiers are instead limited by available C, as suggested by correlated N dissimilation and carbohydrate genes across the gradient (Figure 2, Supplementary Figure S6).
P fertilization effects on decomposition also appear to be contingent on the availability of labile C (Fisk et al., 2015; Nottingham et al., 2015; Poeplau et al., 2016). The strong relationship we observed between inorganic P availability and C turnover (Figure 1) in our rice soils resembles coupling of available P with microbial metabolic rates across land use and global habitat types, including wetlands (Strickland et al., 2010; Hartman and Richardson, 2013). We hypothesized that this variation in C cycling and metabolism arises from underlying shifts in microbial community structure and function, and that coupling of ecosystem biogeochemical cycles reflects metabolic coupling of element use within and among microbial taxa.
Microbial communities and traits linked with stoichiometry and C cycling
Microbial community structure varied with several factors along the soil gradient, including strong independent relationships with soil P stoichiometry (Table 1), in agreement with our second hypothesis. The abundances of several microbial groups were correlated with soil C turnover, including members of the Actinobacteria, Firmicutes, Chloroflexi, some clades of the Acidobacteria and narrower groups of Proteobacteria and Bacteroidetes (Figure 3, Supplementary Figure S7b–c, Supplementary Table S7). Many of these lineages have been previously associated with higher rates of soil C cycling or microbial metabolism (Fierer et al., 2007; Goldfarb et al., 2011; Fierer et al., 2012; Ramirez et al., 2012) potentially reflecting their greater carbohydrate utilization potential (Berlemont and Martiny, 2013; Berlemont et al., 2014).
In our soils, microbial taxa correlated with C turnover rates were enriched in imputed carbohydrate utilization genes while harboring fewer genes for P scavenging (Figure 3), consistent with biogeochemical and metabolic shifts observed along the gradient (Figures 1 and 2). Organisms that were highly correlated with soil C turnover also had greater imputed ribosome gene copy numbers (Figure 3), which are associated with increased growth rates among cultured microbes (Klappenbach et al., 2000; Shrestha et al., 2007; Vieira-Silva and Rocha, 2010; Roller et al., 2016). This key trait also integrates growth rates with cellular demand for N and P (Elser et al., 1996, 2003), underlying functional differentiation and trophic status in higher organisms (Sterner and Elser, 2002; Wright et al., 2004; Arrigo, 2005; Hessen et al., 2013).
Trait differentiation underlies community assembly and C cycling
Tradeoffs in allocation to resource acquisition and growth contribute to metabolic specialization and niche partitioning among organisms (Tilman, 1990; Johnson et al., 2012; Edwards et al., 2013a; Litchman et al., 2015a, b; Díaz et al., 2016). In our data, organisms inferred to use proportionately more carbohydrates had less allocation to P scavenging (Figure 4, Supplementary Figure S8, Supplementary Table S9), consistent with their opposing patterns among OTUs positively or negatively correlated with C turnover (Figure 3), and with shifts in ecosystem C and P cycling in soil biogeochemical and metagenomic data (Figures 1 and 2, Supplementary Figure S6).
Allocation to use of C, N and P substrates differed among high-level taxonomic groups (Figure 4, Supplementary Figure S8,Table 3), in agreement with our third hypothesis, that genomic features and potential utilization of C, N and P substrates would differ characteristically among taxa. Taxonomic patterns in carbohydrate use and P scavenging in our data also appeared broadly similar to those observed in comparative genomic and metagenomic studies (Berlemont and Martiny, 2013; Berlemont et al., 2014; Chai et al., 2014). Deeply rooted partitioning of nutrient allocation could help account for consistent biogeographic patterns in microbial communities (Phillipot et al., 2010; Ramirez et al., 2014) and their responses to fertilization at high taxonomic ranks (Leff et al., 2015; Spohn et al., 2015). Higher taxonomic ranks also accounted for most of the covariation of microbial communities with chemistry in our soils (Supplementary Table S10), consistent with results compared across terrestrial and aquatic ecosystems (Lu et al., 2016).
Differences in resource allocation strategies may underlie patterns of community assembly across environmental gradients (Weber and Deutsch, 2010; Edwards et al., 2013a, b; van Bodegom et al., 2014; Litchman et al., 2015a, b). Our CATS models (Warton et al., 2015) revealed interactions between soil nutrient availability and genomic resource allocation (Figure 4d) that suggest differences in nutrient utilization shape community composition. Genomic allocation to carbohydrate use and N dissimilation both had positive interactions with nitrate concentrations across taxonomic ranks (Figure 4, Supplementary Figure S9), suggesting again that higher denitrification rates (Ye et al., 2016) and genetic potential in our mineral soils (Figure 2) were associated with greater carbohydrate use, analogous to coupled mineral N and carbohydrate use in upland soils (Ramirez et al., 2012; Amend et al., 2015).
Although P scavenging allocation was less clearly related to soil nutrient availability, ribosome copy numbers had significant positive interactions with inorganic P concentrations at each taxonomic rank tested (Figure 4d, Supplementary Figure S9). This result could suggest that inorganic P availability limits the abundance of high ribosome copy number organisms, whose high growth rates and low C use efficiency (Roller et al., 2016) may in part account for greater C turnover in our P-rich mineral soils. This mechanism, if observed elsewhere, could in part account for large-scale relationships between inorganic P availability and microbial metabolic rates across soils (Strickland et al., 2010; Hartman and Richardson, 2013).
Incorporation of functional trait variation can enable community-driven metabolic modeling of nutrient-mediated changes in ecosystem C cycling (Weber and Deutsch, 2010; Bouskill et al., 2012; Litchman et al., 2015a; Martiny et al., 2015), which may be simplified by accounting for tradeoffs among traits (Edwards et al., 2012; Johnson et al., 2012; Litchman et al., 2015b). We tested simplified representations of microbial communities by deriving de novo functional ‘guilds’ of microbes based on similar patterns of imputed C, N and P use. Accounting for differences in carbohydrate allocation (Ccarb:arom) and ribosome copy numbers among these guilds yielded stronger relationships with soil C cycling in our soils than phylogenetic relationships alone (Table 4), while using fewer free parameters (Supplementary Table S11). This finding supports our fourth hypothesis that rates of soil C cycling are linked with the abundance of microbial groups defined by differences in stoichiometric resource allocation, although this awaits confirmation in other ecosystems.
Conclusions
We observed that C turnover across a large soil C:N:P gradient is in part mediated by P availability, reflecting differences in microbial P metabolism related to growth rates and nutrient use. Interactions among ecosystem C, N and P cycling were mirrored by community metabolic potential and appear related to shifts in community structure arising from trait-based environmental filtering of organisms by soil nutrient availability. Our findings suggest that stoichiometric resource allocation and ribosome copy number are key traits that underpin patterns of microbial community structure and function in some soils and may mediate ecosystem C cycling under multiple nutrient limitation. Grouping microbes by their resource allocation patterns improved relationships with soil nutrients and C turnover in the rice paddy soils studied, and the tradeoffs we observed among traits might help to simplify future efforts to assimilate microbial sequence data into ecosystem models.
Data availability
Annotated metagenome sequences are available from the Joint Genome Institute’s IMG database (http://img.jgi.doe.gov) under IDs 3300005873–3300005905. Shotgun metagenome and 16S rRNA data are deposited together in the NCBI Sequence Read Archive, under SRA study numbers SRP099852–SRP099861, SRP099869–SRP099878, SRP099883–SRP099890 and SRP099897–SRP099900.
References
Allen AP, Gillooly JF . (2009). Towards an integration of ecological stoichiometry and the metabolic theory of ecology to better understand nutrient cycling. Ecol Letts 12: 369–384.
Allison SD, Martiny JBH . (2008). Resistance, resilience, and redundancy in microbial communities. Proc Natl Acad Sci 105: 11512–11519.
Amend AS, Martiny AC, Allison SD, Berlemont R, Goulden ML, Lu Y et al. (2015). Microbial response to simulated global change is phylogenetically conserved and linked with functional potential. ISME J 10: 109–118.
Arrigo KR . (2005). Marine microorganisms and global nutrient cycles. Nature 437: 349–355.
Bengtsson-Palme J, Hartmann M, Eriksson KM, Larsson J . (2015). METAXA2: improved identification and taxonomic classification of small and large subunit rRNA in metagenomic data. Mol Ecol Resourc 15: 1403–1414.
Berlemont R, Martiny AC . (2013). Phylogenetic distribution of potential cellulases in bacteria. Appl Environ Microbiol 79: 1545–1554.
Berlemont R, Allison SD, Weihe C, Lu Y, Brodie EL, Martiny JBH et al. (2014). Cellulolytic potential under environmental changes in microbial communities from grassland litter. Front Microbiol 5: 639–639.
Bouskill NJ, Tang J, Riley WJ, Brodie EL . (2012). Trait-based representation of biological nitrification: model development, testing, and predicted community composition. Front Microbiol 3: 364–364.
Bradford MA, Fierer N, Reynolds JF . (2008). Soil carbon stocks in experimental mesocosms are dependent on the rate of labile carbon, nitrogen and phosphorus inputs to soils. Funct Ecol 22: 964–974.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK et al. (2010). QIIME allows analysis of high- throughput community sequencing data. Nat Methods 7: 335–336.
Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N et al. (2012). Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6: 1621–1624.
Cederlund H, Wessén E, Enwall K, Jones CM, Juhanson J, Pell M et al. (2014). Soil carbon quality and nitrogen fertilization structure bacterial communities with predictable responses of major bacterial phyla. Appl Soil Ecol 84: 62–68.
Chai J, Kora G, Ahn T-H, Hyatt D, Pan C . (2014). Functional phylogenomics analysis of bacteria and archaea using consistent genome annotation with UniFam. BMC Evol Biol 14: 207–207.
Cleveland CC, Liptzin D . (2007). C:N:P stoichiometry in soil: is there a ‘Redfield ratio’ for the microbial biomass? Biogeochemistry 85: 235–252.
Craine JM, Morrow C, Fierer N . (2007). Microbial nitrogen limitation increases decomposition. Ecology 88: 2105–2113.
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K et al. (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72: 5069–5072.
Díaz S, Kattge J, Cornelissen JHC, Wright IJ, Lavorel S, Dray S et al. (2016). The global spectrum of plant form and function. Nature 529: 167–171.
Edwards KF, Thomas MK, Klausmeier CA, Litchman E . (2012). Allometric scaling and taxonomic variation in nutrient utilization traits and maximum growth rate of phytoplankton. Limnol Oceanogr 57: 554–566.
Edwards KF, Klausmeier CA, Litchman E . (2013a). A three-way trade-off maintains functional diversity under variable resource supply. Am Nat 182: 786–800.
Edwards KF, Litchman E, Klausmeier CA . (2013b). Functional traits explain phytoplankton community structure and seasonal dynamics in a marine ecosystem. Ecol Lett 16: 56–63.
Elser JJ, Dobberfuhl DR, MacKay NA, Schampel JH . (1996). Organism size, life history, and N:P stoichiometry. Bioscience 46: 674–684.
Elser JJ, Sterner RW, Gorokhova E, Fagan WF, Markow TA, Cotner JB et al. (2000). Biological stoichiometry from genes to ecosystems. Ecol Lett 3: 540–550.
Elser JJ, Acharya K, Kyle M, Cotner J, Makino W, Markow T et al. (2003). Growth rate-stoichiometry couplings in diverse biota. Ecol Lett 6: 936–943.
Espe MB, Kirk E, van Kessel C, Horwath WH, Linquist BA . (2015). Indigenous nitrogen supply of rice is predicted by soil organic carbon. Soil Sci Soc Am J 79: 569–576.
Fanin N, Fromin N, Buatois B, Hättenschwiler S . (2013). An experimental test of the hypothesis of non-homeostatic consumer stoichiometry in a plant litter-microbe system. Ecol Lett 16: 764–772.
Fierer N, Bradford MA, Jackson RB . (2007). Toward an ecological classification of soil bacteria. Ecology 88: 1354–1364.
Fierer N, Lauber CL, Ramirez KS, Zaneveld J, Bradford MA, Knight R . (2011). Comparative metagenomic, phylogenetic and physiological analyses of soil microbial communities across nitrogen gradients. ISME J 6: 1007–1017.
Fierer N, Lauber CL, Ramirez KS, Zaneveld J, Bradford MA, Knight R . (2012). Comparative metagenomic, phylogenetic and physiological analyses of soil microbial communities across nitrogen gradients. ISME J 6: 1007–1017.
Fisk M, Santangelo S, Minick K . (2015). Carbon mineralization is promoted by phosphorus and reduced by nitrogen addition in the organic horizon of northern hardwood forests. Soil Biol Biochem 81: 212–218.
Follows MJ, Dutkiewicz S . (2011). Modeling diverse communities of marine microbes. Annu Rev Mar Sci 3: 427–451.
Franklin O, Hall EK, Kaiser C, Battin TJ, Richter A . (2011). Optimization of biomass composition explains microbial growth-stoichiometry relationships. Am Nat 177: E29–E42.
Freeman C, Ostle N, Kang H . (2001). An enzymic ‘latch’ on a global carbon store. Nature 409: 149–149.
Galperin MY, Makarova KS, Wolf YI, Koonin EV . (2015). Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res 43: D261–D269.
Goldfarb KC, Karaoz U, Hanson CA, Santee CA, Bradford MA, Treseder KK et al. (2011). Differential growth responses of soil bacterial taxa to carbon substrates of varying chemical recalcitrance. Front Microbiol 2: 94–94.
Goll DS, Brovkin V, Parida BR, Reick CH, Kattge J, Reich PB et al. (2012). Nutrient limitation reduces land carbon uptake in simulations with a model of combined carbon, nitrogen and phosphorus cycling. Biogeosciences 9: 3547–3569.
Graham EB, Knelman JE, Schindlbacher A, Siciliano S, Breulmann M, Yannarell A et al. (2016). Microbes as engines of ecosystem function: when does community structure enhance predictions of ecosystem processes? Front Microbiol 7: 214–214.
Griffiths BS, Spilles A, Bonkowski M . (2012). C : N : P stoichiometry and nutrient limitation of the soil microbial biomass in a grazed grassland site under experimental P limitation or excess. Ecol Processes 1: 1–1.
Güsewell S, Gessner MO . (2009). N:P ratios influence litter decomposition and colonization by fungi and bacteria in microcosms. Funct Ecol 23: 211–219.
Hall EK, Maixner F, Franklin O, Daims H, Richter A, Battin T . (2011). Linking microbial and ecosystem ecology using ecological stoichiometry: a synthesis of conceptual and empirical approaches. Ecosystems 14: 261–273.
Hamady M, Lozupone C, Knight R . (2009). Fast UniFrac: facilitating high-throughput phylogenetic analyses of microbial communities including analysis of pyrosequencing and PhyloChip data. ISME J 4: 17–27.
Hartman WH, Richardson CJ . (2013). Differential nutrient limitation of soil microbial biomass and metabolic quotients (qCO2): is there a biological stoichiometry of soil microbes? PLoS One 8: e57127–e57127.
Helton AM, Ardón M, Bernhardt ES . (2015). Thermodynamic constraints on the utility of ecological stoichiometry for explaining global biogeochemical patterns. Ecol Lett 18: 1049–1056.
Hessen DO, Elser JJ, Sterner RW, Urabe J . (2013). Ecological stoichiometry: an elementary approach using basic principles. Limnol Oceanogr 58: 2219–2236.
Ho A, Kerckhof F-M, Luke C, Reim A, Krause S, Boon N et al. (2013). Conceptualizing functional traits and ecological characteristics of methane-oxidizing bacteria as life strategies. Environ Microbiol Rep 5: 335–345.
Huntemann M, Ivanova NN, Mavromatis K, Tripp HJ, Paez-Espino D, Palaniappan K et al. (2015). The standard operating procedure of the DOE-JGI Microbial Genome Annotation Pipeline (MGAP v.4). Standards Genomic Sci 10: 86–86.
Johnson DR, Goldschmidt F, Lilja EE, Ackermann M . (2012). Metabolic specialization and the assembly of microbial communities. ISME J 6: 1985–1991.
Karpinets TV, Greenwood DJ, Sams CE, Ammons JT . (2006). RNA: protein ratio of the unicellular organism as a characteristic of phosphorous and nitrogen stoichiometry and of the cellular requirement of ribosomes for protein synthesis. BMC Biol 4: 30.
Keiblinger KM, Hall EK, Wanek W, Szukics U, Hämmerle I, Ellersdorfer G et al. (2010). The effect of resource quantity and resource stoichiometry on microbial carbon-use-efficiency. FEMS Microbiol Ecol 73: 430–440.
Klappenbach JA, Dunbar JM, Schmidt TM . (2000). rRNA operon copy number reflects ecological strategies of bacteria. Appl Environ Microbiol 66: 1328–1333.
Kögel-Knabner I, Amelung W, Cao Z, Fiedler S, Frenzel P, Jahn R et al. (2010). Biogeochemistry of paddy soils. Geoderma 157: 1–14.
Langille MGI, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 31: 814–821.
Lee HJ, Jeong SE, Kim PJ, Madsen EL, Jeon CO . (2015). High resolution depth distribution of Bacteria, Archaea, methanotrophs, and methanogens in the bulk and rhizosphere soils of a flooded rice. Front Microbiol 6: 639–639.
Leff JW, Jones SE, Prober SM, Barberán A, Borer ET, Firn JL et al. (2015). Consistent responses of soil microbial communities to elevated nutrient inputs in grasslands across the globe. Proc Natl Acad Sci 112: 10967–10972.
Letunic I, Bork P . (2016). Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res 2006: 1–4.
Li J, Wang G, Allison SD, Mayes MA, Luo Y . (2014). Soil carbon sensitivity to temperature and carbon use efficiency compared across microbial-ecosystem models of varying complexity. Biogeochemistry 119: 67–84.
Litchman E, de Tezanos Pinto P, Edwards KF, Klausmeier CA, Kremer CT, Thomas MK . (2015a). Global biogeochemical impacts of phytoplankton: a trait-based perspective. J Ecol 103: 1384–1396.
Litchman E, Edwards KF, Klausmeier CA . (2015b). Microbial resource utilization traits and trade-offs: implications for community structure, functioning, and biogeochemical impacts at present and in the future. Front Microbiol 6: 254–254.
Lu HP, Yeh YC, Sastri AR, Shiah FK, Gong GC, Hsieh CH . (2016). Evaluating community–environment relationships along fine to broad taxonomic resolutions reveals evolutionary forces underlying community assembly. ISME J 10: 2867–2878.
Makino W, Cotner JB . (2004). Elemental stoichiometry of a heterotrophic bacterial community in a freshwater lake: implications for growth-and resource-dependent variations. Aquat Microb Ecol 34: 33–41.
Manzoni S, Trofymow JA, Jackson RB, Porporato A . (2010). Stoichiometric controls on carbon, nitrogen, and phosphorus dynamics in decomposing litter. Ecol Monogr 80: 89–106.
Markowitz VM, Chen IMA, Chu K, Szeto E, Palaniappan K, Pillay M et al. (2014). IMG/M 4 version of the integrated metagenome comparative analysis system. Nucleic Acids Res 42: D568–D573.
Martiny JBH, Jones SE, Lennon JT, Martiny AC . (2015). Microbiomes in light of traits: a phylogenetic perspective. Science 350: aac9323.
McGuire KL, Treseder KK . (2010). Microbial communities and their relevance for ecosystem models: decomposition as a case study. Soil Biol Biochem 42: 529–535.
Melillo JM, Aber JD, Muratore JF . (1982). Nitrogen and lignin control of hardwood leaf litter decomposition dynamics. Ecology 63: 621–626.
Mock T, Daines SJ, Geider R, Collins S, Metodiev M, Millar AJ et al. (2015). Bridging the gap between omics and earth system science to better understand how environmental change impacts marine microbes. Glob Change Biol 22: 61–75.
Mouginot C, Kawamura R, Matulich KL, Berlemont R, Allison SD, Amend AS et al. (2014). Elemental stoichiometry of Fungi and Bacteria strains from grassland leaf litter. Soil Biol Biochem 76: 278–285.
Nottingham AT, Turner BL, Stott AW, Tanner EVJ . (2015). Nitrogen and phosphorus constrain labile and stable carbon turnover in lowland tropical forest soils. Soil Biol Biochem 80: 26–33.
Parikh SJ, Goyne KW, Margenot AJ, Calderón FJ . (2014) Soil Chemical Insights Provided through Vibrational Spectroscopy. USDA Agricultural Research Service: Lincoln, Nebraska, USA.
Phillipot L, Andesson SGE, Battin T, Prosser J, Schimel JP, Whitman WB et al. (2010). The ecological coherence of high bacterial taxonomic ranks. Nat Rev Microbiol 8: 523–529.
Poeplau C, Herrmann AM, Kätterer T . (2016). Opposing effects of nitrogen and phosphorus on soil microbial metabolism and the implications for soil carbon storage. Soil Biol Biochem 100: 83–91.
R Core Team. (2015) R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria.
Ramirez KS, Lauber CL, Knight R, Bradford MA, Fierer N . (2010). Consistent effects of nitrogen fertilization on soil bacterial communities in contrasting systems. Ecology 91: 3463–3470.
Ramirez KS, Craine JM, Fierer N . (2012). Consistent effects of nitrogen amendments on soil microbial communities and processes across biomes. Glob Change Biol 18: 1918–1927.
Ramirez KS, Leff JW, Barberán A, Bates ST, Betley J, Thomas W et al. (2014). Biogeographic patterns in below-ground diversity in New York City’s Central Park are similar to those observed globally. Proc R Soc B Biol Sci 281: 20141988–20141988.
Redfield AC . (1958). The biological control of chemical factors in the environment. Am Sci 46: 205–221.
Reed SC, Yang X, Thornton PE . (2015). Incorporating phosphorus cycling into global modeling efforts: a worthwhile, tractable endeavor. New Phytol 208: 324–329.
Roller BRK, Stoddard SF, Schmidt TM . (2016). Exploiting rRNA operon copy number to investigate bacterial reproductive strategies. Nat Microbiol 1: 16160–16160.
Schimel JP, Schaeffer SM . (2012). Microbial control over carbon cycling in soil. Front Microbiol 3: 1–11.
Shrestha PM, Noll M, Liesack W . (2007). Phylogenetic identity, growth-response time and rRNA operon copy number of soil bacteria indicate different stages of community succession. Environ Microbiol 9: 2464–2474.
Singh BK, Bardgett RD, Smith P, Reay DS . (2010). Microorganisms and climate change: terrestrial feedbacks and mitigation options. Nat Rev Microbiol 8: 779–790.
Sistla SA, Schimel JP . (2012). Stoichiometric flexibility as a regulator of carbon and nutrient cycling in terrestrial ecosystems under change. New Phytol 196: 68–78.
Spohn M, Chodak M . (2015). Microbial respiration per unit biomass increases with carbon-to-nutrient ratios in forest soils. Soil Biol Biochem 81: 128–133.
Spohn M, Treichel NS, Cormann M, Schloter M, Fischer D . (2015). Distribution of phosphatase activity and various bacterial phyla in the rhizosphere of Hordeum vulgare L. depending on P availability. Soil Biol Biochem 89: 44–51.
Sterner RW, Elser JJ . (2002) Ecological Stoichiometry: The Biology of Elements from Molecules to the Biosphere. Princeton University Press: Princeton, NJ, USA.
Strickland MS, Callaham MA, Davies CA, Lauber CL, Ramirez K, Richter DD et al. (2010). Rates of in situ carbon mineralization in relation to land-use, microbial community, and edaphic characteristics. Soil Biol Biochem 42: 260–269.
Tilman D . (1990). Constraints and tradeoffs: toward a predictive theory of competition and succession. Oikos 58: 3–15.
Tremblay J, Singh K, Fern A, Kirton ES, He S, Woyke T et al. (2015). Primer and platform effects on 16S rRNA tag sequencing. Front Microbiol 6: 771–771.
Treseder KK . (2008). Nitrogen additions and microbial biomass: a meta-analysis of ecosystem studies. Ecol Lett 11: 1111–1120.
Treseder KK, Kivlin SN, Hawkes CV . (2011). Evolutionary trade-offs among decomposers determine responses to nitrogen enrichment. Ecol Lett 14: 933–938.
van Bodegom PM, Douma JC, Verheijen LM . (2014). A fully traits-based approach to modeling global vegetation distribution. Proc Natl Acad Sci USA 111: 13733–13738.
Vieira-Silva S, Rocha EPC . (2010). The systemic imprint of growth and its uses in ecological (meta)genomics. PLoS Genet 6: e1000808–e1000808.
Vitousek PM, Porder S, Houlton BZ, Chadwick OA . (2010). Terrestrial phosphorus limitation: mechanisms, implications, and nitrogen-phosphorus interactions. Ecol Appl 20: 5–15.
Vrede T, Dobberfuhl DR, Kooijma SALM, Elser JJ . (2004). Fundamental connections among organism C:N:P stoichiometry, macromolecular composition, and growth. Ecology 85: 1217–1229.
Wang G, Jagadamma S, Mayes MA, Schadt CW, Steinweg JM, Gu L et al. (2015). Microbial dormancy improves development and experimental validation of ecosystem model. ISME J 9: 226–237.
Wang YP, Houlton BZ, Field CB . (2007). A model of biogeochemical cycles of carbon, nitrogen, and phosphorus including symbiotic nitrogen fixation and phosphatase production. Glob Biogeochem Cycles 21: GB1018.
Warton DI, Shipley B, Hastie T . (2015). CATS regression—a model-based approach to studying trait-based community assembly. Methods Ecol Evol 6: 389–398.
Weber TS, Deutsch C . (2010). Ocean nutrient ratios governed by plankton biogeography. Nature 467: 550–553.
Weider WR, Allison SD, Davidson EA, Georgiou K, Hararuk O, He Y et al. (2015). Explicitly representing soil microbial processes in Earth system models. Glob Biogeochem Cycles 29: 1782–1800.
Wright IJ, Reich PB, Westoby M, Ackerly DD, Baruch Z, Bongers F et al. (2004). The worldwide leaf economics spectrum. Nature 428: 821–827.
Yang X, Thornton PE, Ricciuto DM, Post WM . (2014). The role of phosphorus dynamics in tropical forests—a modeling study using CLM-CNP. Biogeosciences 11: 1667–1681.
Ye R, Espe MB, Linquist B, Parikh SJ, Doane TA, Horwath WR . (2016). A soil carbon proxy to predict CH4 and N2O emissions from rewetted agricultural peatlands. Agric Ecosyst Environ 220: 64–75.
Zechmeister-Boltenstern S, Keiblinger KM, Mooshammer M, Penuelas J, Richter A, Sardans J et al. (2015). The application of ecological stoichiometry to plant-microbial-soil organic matter transformations. Ecol Monogr 85: 133–155.
Zhang Z, Li H, Hu J, Li X, He Q, Tian G et al. (2015). Do microorganism stoichiometric alterations affect carbon sequestration in paddy soil subjected to phosphorus input? Ecol Appl 25: 866–879.
Acknowledgements
This project was funded by the DOE Early Career Research Program, grant no. KP/CH57/1, and conducted by the US Department of Energy Joint Genome Institute, a DOE Office of Science User Facility, supported under contract no. DE-AC02-05CH11231. This material is also based on work supported by the National Institute of Food and Agriculture, US Department of Agriculture, under award number: 2011-67003-30371. We thank Nick Bouskill for productive conversations and comments and two anonymous reviewers for helpful suggestions to improve the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Hartman, W., Ye, R., Horwath, W. et al. A genomic perspective on stoichiometric regulation of soil carbon cycling. ISME J 11, 2652–2665 (2017). https://doi.org/10.1038/ismej.2017.115
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2017.115
This article is cited by
-
Community metagenomics reveals the processes of nutrient cycling regulated by microbial functions in soils with P fertilizer input
Plant and Soil (2023)
-
Comparison in structure and predicted function of epiphytic bacteria on Neopyropia yezoensis and Neopyropia katadae
Journal of Oceanology and Limnology (2023)
-
Tree functional traits, forest biomass, and tree species diversity interact with site properties to drive forest soil carbon
Nature Communications (2022)
-
Mechanism that allows manno-oligosaccharide to promote cellulose degradation by the bacterial community and the composting of cow manure with straw
Environmental Science and Pollution Research (2022)
-
Uncovering multi-faceted taxonomic and functional diversity of soil bacteriomes in tropical Southeast Asian countries
Scientific Reports (2021)
