
Abstract
Reticulon 4 receptor (RTN4R) plays an essential role in regulating axonal regeneration and plasticity in the central nervous system through the activation of rho kinase, and is located within chromosome 22q11.2, a region that is known to be a hotspot for schizophrenia (SCZ) and autism spectrum disorder (ASD). Recently, rare variants such as copy-number variants and single-nucleotide variants have been a focus of research because of their large effect size associated with increased susceptibility to SCZ and ASD and the possibility of elucidating the pathophysiology of mental disorder through functional analysis of the discovered rare variants. To discover rare variants with large effect size and to evaluate their role in the etiopathophysiology of SCZ and ASD, we sequenced the RTN4R coding exons with a sample comprising 370 SCZ and 192 ASD patients, and association analysis using a large number of unrelated individuals (1716 SCZ, 382 ASD and 4009 controls). Through this mutation screening, we discovered four rare (minor allele frequency <1%) missense mutations (R68H, D259N, R292H and V363M) of RTN4R. Among these discovered rare mutations, R292H was found to be significantly associated with SCZ (P=0.048). Furthermore, in vitro functional assays showed that the R292H mutation affected the formation of growth cones. This study strengthens the evidence for association between rare variants within RTN4R and SCZ, and may shed light on the molecular mechanisms underlying the neurodevelopmental disorder.
Similar content being viewed by others

Introduction
Schizophrenia (SCZ) is a devastating psychiatric disorder that is characterized by hallucinations, delusions and cognitive deficits, and which causes tremendous societal burdens.1 The lifetime prevalence of SCZ is ~1% in the general population. The heritability of SCZ is up to 80%, making this condition a target for human genetics research.2, 3 Recent large-scale genome-wide association analysis,4 whole-exome sequencing5, 6 and copy-number variant analysis7 with SCZ samples have revealed that deleterious rare gene variants such as single-nucleotide variants (SNVs) and copy-number variants exert significantly larger effects than common single-nucleotide polymorphisms (SNPs). Furthermore, rare SNVs, discovered from sequencing of susceptibility genes, may have large effect sizes and account for a part of the heritability of SCZ, and could contribute to an understanding of the etiopathophisiology of neurodevelopmental disease through further functional assays.8, 9, 10, 11 Recently, disturbances of neuronal connectivity in both local neural circuits and broad networks of interconnected brain areas have been implicated in SCZ12, 13 and white matter, myelin and oligodendrocyte have received increasing attention in terms of their potential role in the etiopathophysiology of SCZ.14, 15 Thus, the sequencing of myelin-related genes might be a hopeful method for uncovering the etiopathophysiology of SCZ.
The Reticulon 4 receptor (RTN4R) encodes the RTN4 receptor, which is a known receptor subunit for RTN4 that is one of the most potent myelin-associated inhibitor of axon regeneration and structural plasticity in the central nervous system.13, 16 RTN4R is considered to be a promising candidate gene for SCZ and ASD because RTN4R is located at chr22q11.2, which is a hotspot locus for SCZ and ASD.17 Carriers with chr22q11.2 deletion exhibit a 20–30% prevalence for SCZ, and this deletion is one of the highest known risk factors for SCZ.17, 18 RTN4R is known to form a co-receptor complex with other proteins such as immunoglobulin domain-containing protein (LINGO1),19 p75, and tumor necrosis factor receptor orphan Y (TROY),13 thereby activating the Ras homolog gene family, member A (RhoA).20 This activation initiates a cascade of intracellular molecular events that results in the destabilization of the actin cytoskeleton and leads to the collapse of growth cones, preventing further axonal growth and inhibiting myelination.16 Furthermore, during development, there is evidence that RTN4R is involved in limiting the critical period of experience-driven plasticity21 and restricts synapse development in the hippocampus.22 Thus, RTN4-related signaling may act to stabilize brain wiring both in adulthood and during development and may relate to both learning and memory.23 Moreover, mice lacking RTN4R exhibit reduced working memory function, consistent with an end phenotype of SCZ.24 In addition, the SNP (rs701428) in RTN4R is associated with the internal capsule of SCZ based on a diffusion tensor imaging study of the brain.25
Considering that RTN4R is strongly related to the neurodevelopment and is located at 22q11.2, we hypothesized that variants in RTN4R possibly contribute to ASD and SCZ susceptibility. Although there have been studies that have focused on SNVs of RTN4R,24, 26 there have been no study that has detected SNVs with a large effect size on SCZ and that focused on SNVs in RTN4R in a Japanese SCZ population. Furthermore, it has reported that the common variants of the RTN4R had association with Japanese SCZ.27 Therefore, in this study, in order to discover novel rare RTN4R mutations with large effect size and to evaluate the pathogenesis of the discovered mutations, we sequenced the RTN4R coding exons using SCZ and ASD samples, and performed the association analysis and in vitro functional assays of the variants that could have large effects. Through this study, we elucidated that a novel rare variant R292H in RTN4R affects growth cone formation and possibly contributes to SCZ susceptibility.
Materials and methods
Participants
Two independent sample sets were used in this study. The first set, which comprised 370 SCZ patients (mean age=49.7±14.7 years; males=52.9%) and 192 ASD patients (mean age=16.3±8.36 years; males=77.6%), was sequenced for rare variants of RTN4R. The second, larger set, which comprised 1716 SCZ patients (mean age=47.6±15.2 years; males=52.5%), 382 ASD patients (mean age=19.6±10.7 years; males=77.7%) and 4009 controls (mean age=44.1±14.2 years; males=45.5%), was used for association analysis of selected variants detected in the first set. All of the cases were included if they met DSM-5 criteria for SCZ and ASD. In addition, the patient’s capacity to consent was confirmed by a family member when needed. Subjects with a legal measure of reduced capacity were excluded. Control subjects were healthy volunteers from the general public who had no history of mental disorders, based on questionnaire responses from the subjects themselves during the sample inclusion step, and based on an unstructured diagnostic interview done by an experienced psychiatrist during the blood collection step; this was ascertained during face-to-face interviews where subjects were asked whether they had suffered episodes of depression, mania or psychotic experiences or if they had received treatment for any psychotic disorders. This study protocol was approved by the Ethics Committees of the Nagoya University Graduate School of Medicine and other participating institutes and hospitals. The study was conducted in accordance with the established ethical standards of all institutions.
Mutation screening
Genomic DNA was extracted from whole blood or saliva using the Qiagen QIAamp DNA blood kit or tissue kit (Qiagen, Hilden, Germany). Custom amplification primers were designed to span coding exons and flanking intron regions of the selected genes (transcription ID of RTN4R: ENST00000043402 from ensemble database; human reference sequence NCBI built 37) using the Ion AmpliSeq Designer (Thermo Fisher Scientific, Waltham, MA, USA). Sample amplification and equalization were achieved using Ion AmpliSeq Library Kits 2.0 and the Ion Library Equalizer Kit, respectively (Thermo Fisher Scientific). Amplified sequences were ligated with Ion Xpress Barcode Adapters (Thermo Fisher Scientific). Emulsion PCR and subsequent enrichment were performed using the Ion OneTouch Template Kit v2.0 on Ion OneTouch 2 and Ion OneTouch ES, respectively (Thermo Fisher Scientific). The final product was then sequenced on the Ion PGM sequencing platform (Thermo Fisher Scientific). Raw data output from the sequencer with the default setting; call quality ⩾20, read depth ⩾10 was uploaded to the Torrent Server (Life Technologies, Carlsbad, CA, USA) for variant calling with NCBI GRCh37 as a reference. The resulting VCF files were analyzed by Ingenuity Variant Analysis (Qiagen) for annotation and visualization.
Prioritization
Nonsense mutations, missense mutations, small insertions/deletions and canonical splicing site variations with an allele frequency of <1% were selected from the annotated data. The variants selected through the above-mentioned filtering were validated by Sanger sequencing. About the missense mutations, we chose the variants located in a functional domain or motif of the protein, according to the Human Protein Reference Database (http://www.hprd.org)
All mutations were evaluated in silico for possible structural and functional consequences using the following tools: (1) localization of a functional domain or motif of the protein was based on the Human Protein Reference Database (http://www.hprd.org/indexhtml) and existing literature;24 (2) evolutionary conservation was assessed with the Evola var. 7.5 (http://www.h-invitational.jp/evola/search.html); and (3) prediction of deleterious effects was performed by in silico analytic methods (Polyphen-2[ref. 28](http://genetics.bwh.harvard.edu/pph2/) and SIFT28(http://sift.jcvi.org/)). To investigate the association of discovered rare variants with susceptibility to neuropsychiatric disorders, we performed the association analysis with the further prioritized variants based on the following criteria: (1) novel, not documented in the NCBI dbSNP database (Build 137) (http://www.ncbi.nlm.nih.gov/SNP/), the 1000 Genomes Project. (http://www.1000genomes.org/), the Exome Variant Server of the NHLBI GO Exome Sequencing Project (ESP6500SI-V2) (http://evs.gs.washington.edu/EVS/), the Human Genetic Variation Database (HGVD) of Japanese genetic variation consortium (http://www.genome.med.kyoto-u.ac.jp/SnpDB) or the Exome Aggregation Consortium (ExAC) (http://exac.broadinstitute.org).
Association analysis
Custom TaqMan SNP genotyping assays were designed and ordered from Applied Biosystems (Foster City, CA, USA). Allelic discrimination analysis was performed on an ABI PRISM 7900HT Sequence Detection System (Applied Biosystems). Differences in allele and genotype frequencies of the mutations were compared between SCZ patients/controls and ASD patients/controls using Fisher’s exact test (one-tailed), with a threshold of significance set at P<0.05. Statistical calculations were performed using SPSS v21 (SPSS, Armonk, NY, USA). We performed power analysis using the Genetic Power Calculator (http://pngu.mgh.harvard.edu/purcell/gpc/) based on the multiplicative model.
Plasmid constructs and RTN4R mutagenesis
For expression cloning of RTN4R-EGFP, the Myc tag sequence of the pSecTag2-Hygro vector (Invitrogen, Carlsbad, CA, USA) was replaced with EGFP. The cDNA of human RTN4R was amplified from pENTR221-Hs RTN4R (IMAGE clone 100005521), and was inserted into the Kpn1-Xho1 site of pSecTag2-Hygro by using the signal peptide of pSecTag2. All site-specific mutagenesis experiments were carried out using the KOD-Plus-Mutagenesis Kit (TOYOBO, Shiga, Japan), using the pSecTag2-Hygro-RTN4R vector as a template. The following primers were used for mutagenesis: R292H, 5′-CACCTGGCTGGCCGTGACCTCAAAC-3′, 5′-CGAGGTGCCCTGCAGCCTCCCGCAA-3′ R119W, 5′-TGGTCTGTGGACCCTGCCACATTCC-3′, 5′-GGACCTCAGCGATAATGCACAGCTC-3′, R196H, 5′-CACGCCTTCCGTGGGCTGCACAGCC-3′, 5′- CAACCGCATCTCCAGCGTGCCCGAG-3′. We also constructed GST-LINGO1 expressing plasmid. The cDNA encoding full-length of human LINGO1 was cloned from a human hippocumpus cDNA library (Clontech, Mountain View, CA, USA) and into pEF-BOS-GST vector.29
GST-binding assay
Semi-confluent HEK293FT cells (Thermo Fisher Scientific) were transfected with RTN4R-GFP-related plasmids and GST-LINGO1 expressing plasmid using Lipofectamine 3000 (Thermo Fisher Scientific). After 48 h, the transfected cells were harvested and lysed in 1 ml lysis buffer (50 mM HEPES, pH 7.5, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 1% Triton X-100 and 10% glycerol) on ice19 Glutathione sepharose beads were incubated with the cell lysates for 1 h at 4 °C and the beads were subsequently washed three times with lysis buffer. After washing, the beads were resuspended with sodium dodecyl sulfate polyacrylamide gel electrophoresis sample buffer, and the bound glutathione S-transferase (GST) and green fluorescent protein (GFP) fusion proteins were subjected to immunoblot analysis using the antibodies as indicated in figure.
Neuronal culture and transfections
For dissociated retinal cell cultures, E5.5 chicks were used due to the reduced transfection efficiency in later embryonic period (that is, E8). The eyes were enucleated, and the retinas were dissected and incubated at 37 °C for 30 min in a digestion solution containing papain (16.5 U ml−1; Worthington Biochemical, Lakewood, NJ, USA), DNase (0.5 mg ml−1; Sigma-Aldrich, St. Louis, MO, USA), and L-cysteine (0.3 g ml−1, Sigma) in a phosphate-buffered solution (PBS). The cells were then rinsed with DMEM containing 10% FBS and centrifuged at 1000 r.p.m. for 5 min. The cell suspension was passed through a cell strainer (70 mm; BD Falcon, Corning, NY, USA) to remove cell debris. After centrifugation, the cells were carefully resuspended in DMEM supplemented with B27 (1: 50; Invitrogen), and were then subjected to nucleofection. For nucleofection, the cells were washed and resuspended in room temperature Chick Nucleofector Solution (Lonza, Basel, Switzerland). The cell–nucleofector solution complex (100 μl) and the indicated plasmids (3 μg) were then gently mixed and transferred into a cuvette, followed by nucleofection using the nucleofector program G-013 (Lonza).
Growth cone collapse assay
Three-microliter drops of RTN4-Fc or Fc (R&D Systems, Minneapolis, MN, USA) were spotted and dried on poly-L-lysine-coated dishes. The dishes were then rinsed and coated with 10 μg μl−1 laminin before addition of dissociated chick retinal cells. The cells were fixed with 0.5% glutaraldehyde for 1 h at room temperature, and treated with PBS containing 5% BSA and 0.1% Triton X-100. The cells were then incubated overnight with anti-GFP antibody (Invitrogen) and rhodamine-phalloidin (Invitrogen) at 4 °C, followed by incubation with anti-mouse IgG antibody conjugated with Alexa Fluor 488 (Invitrogen) for 1 h at room temperature. Finally, the signal was analyzed using an Olympus BX53 microscope (Olympus, Tokyo, Japan). The average of three experiment was measured, and at least 100 cells were counted per experiment.
Modeling of the 3D complex structure of RTN4R and LINGO1
The 3D complex structure of RTN4R and LINGO1 was modeled by superimposing their monomeric 3D structures (PDBIDs: 1p8t and 4oqt, respectively) on the template 3D structure of the human SLITRK1 and PTPRD complex (PDBID: 4rca). The template structure was found with the help of the HOMCOS server30 by the condition that the 292-nd site of RTN4R became an interface for other proteins. The sequence of the SLITRK1 protein is similar to that of RTN4R, with 33.5% sequence identity, and both the LINGO1 and PTPRD proteins have an immunoglobulin (Ig)-like domain with weak sequence similarity (sequence identity=19.5%). The program MATRAS31 was used for the superimposition. The model was then refined using three programs: MODELLER,32 UCSF Chimera33 and myPresto.34 Details of the modeling procedure are provided in the Supplementary Methods and Supplementary Figure S1.
Results
Mutation screening and prioritization
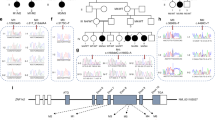
In order to elucidate the association of rare SNVs in RTN4R with the pathophysiology of SCZ and ASD, we sequenced RTN4R coding regions using 370 SCZ and 192 ASD. The resulting nucleotide sequence data have been deposited in the DNA Data Bank of Japan (DDBJ) databases (http://www.ddbj.nig.ac.jp) under the accession number DRA004490. Through this sequencing, we identified four rare missense mutations (MAF<1%) that were validated by Sanger sequencing (Figures 1a–c). We discovered all of the missense mutations within exon 2 (Figures 1a and b). The R292H mutation was not documented in several databases (Table 1). Results obtained from evolutionary conservation analysis of the discovered rare missense mutations are shown in Figure 1d.

The results of mutation screening. (a) RTN4R gene structure based on ENST00000040608; the protein-coding exons sequenced in this study are indicated by yellow box. Untranslated regions (UTR) are indicated by gray box. Four rare missense mutations were discovered in exon 2. (b) RTN4R protein structure (473 amino acids). N-terminal (NT) leucine-rich repeat (LRR) domain (green box), eight LRR domains, CT-LRR domain, a stalk domain and GPI anchorage site (purple box) are contained in RTN4R. R68H, D259N and R292H are located in LRR domain. V363M is located in the Stalk domain. LRR domain is ligand-binding regions such as RTN4, MAG, OMGP and LGI1.35 Stalk domain is the interaction site for co-receptor p75NTR of TROY and is needed for RhoA activation.36 Variants discovered in this study are indicated in square box. Previously published missense mutations24, 26, 37, 38, 39 associated with schizophrenia (SCZ) are underlined. (c) Results of the Sanger sequence showing the discovered rare missense mutations. The mutated sites are indicated by the arrows. (d) The results of evolutionary conservation analysis.
Association analysis
Among the four discovered mutations, R292H was assumed to have a high effect on SCZ based on the following findings; (1) two unrelated cases in the 370 SCZ samples had the R292H mutation, (2) this mutation was not registered in several public databases (Table 1), (3) R292H was located in the RTN4R ligand-binding domain (Figure 1b), which, following ligand binding initiates a cascade of intracellular molecular events that result in destabilization of the actin cytoskeleton and lead to the collapse of growth cones.16 We therefore performed genetic association analysis using RTN4R-R292H. For our SCZ sample of cases (N=2086), we computed a statistical power of >80% based on the following parameters: disease prevalence of 0.01, observed rare allele frequency of 0.0027 (Table 1), odds ratio (OR) for dominant effect of ⩾2.3 and type I error rate of 0.05. The result of association analysis of RTN4R-R292H is shown in Table 2. A marginally significant association between SCZ and R292H was observed (OR=3.9, P=0.048). The clinical features of the six SCZ cases with RTN4R-R292H are shown in Table 3.
Functional analysis of RTN4R-R292H
We considered that the RTN4R-R292H mutation might have strong relations to the pathogenesis of SCZ by affecting RTN4R function from the following findings: (1) R292H was marginally associated with SCZ, (2) according to a previous study,24 SNVs in RTN4R that are located at the RTN4 binding domain (R119W and R196H) inhibited growth cone collapse, (3) the R292H mutation discovered in this study was also located in the ligand-binding site. Therefore, to examine the effect of R292H on neurodevelopment, a growth cone collapse assay was performed in response to the myelin ligand; RTN4, which requires RTN4R expression. We first generated RTN4R mutant plasmids (R292H, R119W and R196H) and examined their subcellular localization in HEK293 cells. There were no obvious differences in subcellular localization between RTN4R wild type (WT) and the mutants (Supplementary Figure S2). We then investigated the effects of these mutants on growth cone collapse in chick retinal cells (Figure 2a). These cells were used because it has been reported that embryonic chick retina show weak expression of RTN4R.40 Overexpression of RTN4R-R292H in the E5.5 chick retinal neurons significantly decreased growth cone collapse compared with that of RTN4R-WT, when the cells were challenged with the RTN4R ligand RTN4. Consistent with a previous study, RTN4R-R119W,24 which is also in the RTN4 ligand-binding site, also significantly decreased RTN4-induced growth cone collapse compared with RTN4R-WT (Figure 2b).

Effect of RTN4R mutants on growth cone collapse. (a) Representative images of growth cone in chick retinal cells. The cells were transfected with indicated plasmid. GFP is used as mock-transfected control. Phalloidin is used as an actin marker. Scale bar, 10 μm. (b) The result of analysis of growth cone collapse. Chick-dissociated retinal neurons transfected with the indicated plasmids encoding EGFP-RTN4R WT or mutants were cultured in the presence or absence of the RNT4R ligand RTN4. The cells were stained with anti-GFP and rhodamine-conjugated phalloidin. Error bars represent ±s.e. n=6, **P<0.01, *P<0.05.
Based on in silico 3D protein structure analysis, we predicted that R292 could be located at the site of interactions between RTN4R and LINGO1 (Figure 3a, Supplementary Methods, and Supplementary Figure S1), which is also an SCZ candidate gene and is known to build a receptor complex with RTN4R and regulate myelination.41, 42 RTN4R-R292 is predicted to form a hydrogen bond with LINGO1-Q404 (Figure 3b). RTN4R-R292H is predicted to be located too far from LINGO1-Q404 for the formation of hydrogen bonds (Figure 3c). We discovered a reduced interaction of LINGO1 with RTN4R-R292H compared to WT (Figure 3d) by the GST-binding assay as our in silico structural analysis predicted.

The predicted 3D model of RTN4R–LINGO1 complex and GST-binding assay. (a) The predicted 3D model of RTN4R (blue) and LINGO1 (red); R292 is located in the yellow circle. (b) Enlarged view around R292; RTN4R-R292 is predicted to form a hydrogen bond with LINGO1-Q404. (c) An enlarged view around H292 of the predicted R292H-mutated structure; the RTN4R-H292 is predicted to be located too far from LINGO1-Q404 for the formation of hydrogen bonds. (d) The GST-binding assay using GST-LINGO1 as a bait protein and GFP-tagged RTN4R WT, R119W, R196W and R292H as pray proteins. Input and bound proteins were detected on immunoblots probed with an anti-GFP or anti-GST antibodies. Immunoblot was detected by using Odyssey Clx (LI-COR, USA) and detected a reduced interaction of LINGO1 with RTN4R-R292H compared to WT.
Discussion
We performed mutation sequencing of RTN4R coding regions as a candidate gene for SCZ and ASD. Through this study, four missense mutations in RTN4R (Table 1a) were discovered. Among four discovered mutations, RTN4R-R292H was not found in several existing databases but detected in two SCZ cases after the mutations screening using 370 SCZ. Through association analysis, a marginally significant association (OR=3.9, P=0.048) was detected between SCZ and RTN4R-R292H in a sample comprising 6095 unrelated individuals. This result is consistent with the recent genetic studies using large samples of neurodevelopmental disorders; deleterious rare mutations with large effect size are highly enriched in SCZ and ASD.18
Furthermore, we found a possible biological effect of RTN4R-R292H by investigating growth cone collapse of chick-dissociated retinal neurons induced by a myelin ligand (RTN4). A previous report also suggested that a decrease in myelin-induced growth cone collapse by SNVs in RTN4R was related to SCZ susceptibility.24 The possible mechanisms by which the RTN4R-R292H mutation induced reduced growth cone collapse may involve changes in the following: (1) interaction of RTN4R with ligands including RTN4 that are required for myelin-mediated neural development;43 (2) RhoA signal transduction due to alteration of ligand-induced conformational changes of the RTN4 receptor; and/or (3) co-receptor association with LINGO1, which is also an SCZ candidate gene and one of the components of RTN4R signaling complex and regulate myelination activating RhoA signaling,19 predicted that R292 could be located at the site of interactions between RTN4R and LINGO1 (Figure 3a, Supplementary Methods and Supplementary Figure S1), and which is also an SCZ candidate gene that is known to build a receptor complex with RTN4R and regulate myelination,41, 42 based on in silico 3D protein structure analysis. We discovered a reduced interaction of LINGO1 with RTN4R-R292H compared to WT (Figure 3d) by the GST-binding analysis as our in silico structural analysis predicted. Through these genetic and functional studies, considering that RTN4R is located at chr22q11.2, which is a hotspot locus for SCZ and ASD,17 RTN4R-R292H could be related to the etiopathological role of SCZ, because the pathophysiology of SCZ is believed to be that of aberrant conditions of neurodevelopment.43
We could get the DNA from the daughter of Subject 2 who was diagnosed with SCZ, and showed that she was also an RTN4R-R292H carrier (Supplementary Figure S4). However, there seems not to share any common clinical information in the six cases with RTN4R-R292H (Table 3, Supplementary Figure S4). Given the fact that SCZ is a disorder of complex genetics with an environmental component, it is not surprising that RTN4R-R292H does not fully reproduce the disease. Functional change of RTN4R-R292H throughout neurodevelopment could possibly increase the SCZ risk.
Several limitations should be discussed when interpreting the results of this study. Firstly, our sequencing did not cover the several potentially informative regions of RTN4R, including untranslated regions and intronic regions. Indeed, it has recently been reported that de novo synonymous mutations in the gene SETD1A could contribute to the genetic etiology of SCZ.44 Secondly, due to limited access to the detailed clinical phenotypes of the subject and subjects’ family members and control subjects with RTN4R-R292H, and to lack of DNA samples from subjects’ family, we could not fully explore the impact of the novel variants segregation. Third, we only discovered a reduction of growth cone collapse by RTN4R-R292H with chick retinal neurons in E5.5. Due to the reduced transfection efficiency in later embryonic period (that is, E8), we could not assess effects of variant in terms of pathophysiological mechanism based on dominant negative or gain-of-function model during the high expression of RTN4R (that is, E13). Finally, the mechanisms of how R292H leads to pathophysiology of the SCZ are left unanswered. Therefore, the potential effect of RTN4R-R292H on cortical neurons and on the RhoA signaling should be investigated in future studies.
In conclusion, individuals with the RTN4R-R292H might increase susceptibility to developing SCZ. Furthermore, we revealed the biological effect of inhibiting myelin-mediated neurite sprouting by the mutation of RTN4R-R292H. This study strengthens the evidence the rare variants in RTN4R to the etiopathological role of SCZ. In future research involving a more comprehensive evaluation of RTN4R, a study with a much larger sample size, and that examines RTN4R-related pathway genes (for example; LINGO1, RTN4, RTN4R and MAG) will be needed.
References
Collins PY, Patel V, Joestl SS, March D, Insel TR, Daar AS et al. Grand challenges in global mental health. Nature 2011; 475: 27–30.
Sullivan PF, Kendler KS, Neale MC . Schizophrenia as a complex trait: evidence from a meta-analysis of twin studies. Arch Gen Psychiatry 2003; 60: 1187–1192.
Lichtenstein P, Yip BH, Bjork C, Pawitan Y, Cannon TD, Sullivan PF et al. Common genetic determinants of schizophrenia and bipolar disorder in Swedish families: a population-based study. Lancet 2009; 373: 234–239.
Lee SH, Ripke S, Neale BM, Faraone SV, Purcell SM, Perlis RH et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet 2013; 45: 984–994.
De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014; 515: 209–215.
Purcell SM, Moran JL, Fromer M, Ruderfer D, Solovieff N, Roussos P et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature 2014; 506: 185–190.
Malhotra D, Sebat J . CNVs: harbingers of a rare variant revolution in psychiatric genetics. Cell 2012; 148: 1223–1241.
Kimura H, Tsuboi D, Wang C, Kushima I, Koide T, Ikeda M et al. Identification of rare, single-nucleotide mutations in NDE1 and their contributions to schizophrenia susceptibility. Schizophr Bull 2015; 41: 744–753.
Hamilton PJ, Campbell NG, Sharma S, Erreger K, Herborg Hansen F, Saunders C et al. De novo mutation in the dopamine transporter gene associates dopamine dysfunction with autism spectrum disorder. Mol Psychiatry 2013; 18: 1315–1323.
Timms AE, Dorschner MO, Wechsler J, Choi KY, Kirkwood R, Girirajan S et al. Support for the N-methyl-D-aspartate receptor hypofunction hypothesis of schizophrenia from exome sequencing in multiplex families. JAMA Psychiatry 2013; 70: 582–590.
Durand CM, Perroy J, Loll F, Perrais D, Fagni L, Bourgeron T et al. SHANK3 mutations identified in autism lead to modification of dendritic spine morphology via an actin-dependent mechanism. Mol Psychiatry 2012; 17: 71–84.
Davis KL, Stewart DG, Friedman JI, Buchsbaum M, Harvey PD, Hof PR et al. White matter changes in schizophrenia: evidence for myelin-related dysfunction. Arch Gen Psychiatry 2003; 60: 443–456.
Yiu G, He Z . Glial inhibition of CNS axon regeneration. Nature reviews Neuroscience 2006; 7: 617–627.
Carletti F, Woolley JB, Bhattacharyya S, Perez-Iglesias R, Fusar Poli P, Valmaggia L et al. Alterations in white matter evident before the onset of psychosis. Schizophr Bull 2012; 38: 1170–1179.
White T, Magnotta VA, Bockholt HJ, Williams S, Wallace S, Ehrlich S et al. Global white matter abnormalities in schizophrenia: a multisite diffusion tensor imaging study. Schizophr Bull 2011; 37: 222–232.
Schwab ME . Functions of Nogo proteins and their receptors in the nervous system. Nat Rev Neurosci 2010; 11: 799–811.
Karayiorgou M, Simon TJ, Gogos JA . 22q11.2 microdeletions: linking DNA structural variation to brain dysfunction and schizophrenia. Nat Rev Neurosci 2010; 11: 402–416.
Schneider M, Debbane M, Bassett AS, Chow EW, Fung WL, van den Bree M et al. Psychiatric disorders from childhood to adulthood in 22q11.2 deletion syndrome: results from the International Consortium on Brain and Behavior in 22q11.2 Deletion Syndrome. Am J Psychiatry 2014; 171: 627–639.
Mi S, Lee X, Shao Z, Thill G, Ji B, Relton J et al. LINGO-1 is a component of the Nogo-66 receptor/p75 signaling complex. Nat Neurosci 2004; 7: 221–228.
Nash M, Pribiag H, Fournier AE, Jacobson C . Central nervous system regeneration inhibitors and their intracellular substrates. Mol Neurobiol 2009; 40: 224–235.
McGee AW, Yang Y, Fischer QS, Daw NW, Strittmatter SM . Experience-driven plasticity of visual cortex limited by myelin and Nogo receptor. Science 2005; 309: 2222–2226.
Wills ZP, Mandel-Brehm C, Mardinly AR, McCord AE, Giger RJ, Greenberg ME . The nogo receptor family restricts synapse number in the developing hippocampus. Neuron 2012; 73: 466–481.
Lee H, Raiker SJ, Venkatesh K, Geary R, Robak LA, Zhang Y et al. Synaptic function for the Nogo-66 receptor NgR1: regulation of dendritic spine morphology and activity-dependent synaptic strength. J Neurosci 2008; 28: 2753–2765.
Budel S, Padukkavidana T, Liu BP, Feng Z, Hu F, Johnson S et al. Genetic variants of Nogo-66 receptor with possible association to schizophrenia block myelin inhibition of axon growth. J Neurosci 2008; 28: 13161–13172.
Perlstein MD, Chohan MR, Coman IL, Antshel KM, Fremont WP, Gnirke MH et al. White matter abnormalities in 22q11.2 deletion syndrome: preliminary associations with the Nogo-66 receptor gene and symptoms of psychosis. Schizophr Res 2014; 152: 117–123.
Sinibaldi L, De Luca A, Bellacchio E, Conti E, Pasini A, Paloscia C et al. Mutations of the Nogo-66 receptor (RTN4R) gene in schizophrenia. Hum Mutat 2004; 24: 534–535.
Jitoku D, Hattori E, Iwayama Y, Yamada K, Toyota T, Kikuchi M et al. Association study of Nogo-related genes with schizophrenia in a Japanese case-control sample. Am J Med Genet B Neuropsychiatr Genet 2011; 156B: 581–592.
Kumar P, Henikoff S, Ng PC . Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 2009; 4: 1073–1081.
Mizushima S, Nagata S . pEF-BOS, a powerful mammalian expression vector. Nucleic Acids Res 1990; 18: 5322.
Kawabata T . HOMCOS: an updated server to search and model complex 3D structures. J Struct Funct Genomics 2016; 17: 83–99.
Kawabata T, Nishikawa K . Protein structure comparison using the markov transition model of evolution. Proteins 2000; 41: 108–122.
Sali A, Blundell TL . Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 1993; 234: 779–815.
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 2004; 25: 1605–1612.
Fukunishi YMY, Nakamura H . The filling potential method: a method for estimating the free energy surface for protein−ligand docking. J Phys Chem B 2003; 107: 13201.
He XL, Bazan JF, McDermott G, Park JB, Wang K, Tessier-Lavigne M et al. Structure of the Nogo receptor ectodomain: a recognition module implicated in myelin inhibition. Neuron 2003; 38: 177–185.
Wang KC, Kim JA, Sivasankaran R, Segal R, He Z . P75 interacts with the Nogo receptor as a co-receptor for Nogo, MAG and OMgp. Nature 2002; 420: 74–78.
Hsu R, Woodroffe A, Lai WS, Cook MN, Mukai J, Dunning JP et al. Nogo receptor 1 (RTN4R) as a candidate gene for schizophrenia: analysis using human and mouse genetic approaches. PLoS ONE 2007; 2: e1234.
Lazar NL, Singh S, Paton T, Clapcote SJ, Gondo Y, Fukumura R et al. Missense mutation of the reticulon-4 receptor alters spatial memory and social interaction in mice. Behav Brain Res 2011; 224: 73–79.
Thomas RA, Ambalavanan A, Rouleau GA, Barker PA . Identification of genetic variants of LGI1 and RTN4R (NgR1) linked to schizophrenia that are defective in NgR1-LGI1 signaling. Mol Genet Genomic Med 2016; 4: 447–456.
Fournier AE, GrandPre T, Strittmatter SM . Identification of a receptor mediating Nogo-66 inhibition of axonal regeneration. Nature 2001; 409: 341–346.
Mi S, Miller RH, Lee X, Scott ML, Shulag-Morskaya S, Shao Z et al. LINGO-1 negatively regulates myelination by oligodendrocytes. Nat Neurosci 2005; 8: 745–751.
Lee X, Yang Z, Shao Z, Rosenberg SS, Levesque M, Pepinsky RB et al. NGF regulates the expression of axonal LINGO-1 to inhibit oligodendrocyte differentiation and myelination. J Neurosci 2007; 27: 220–225.
Willi R, Schwab ME . Nogo and Nogo receptor: relevance to schizophrenia? Neurobiol Dis 2013; 54: 150–157.
Takata A, Ionita-Laza I, Gogos JA, Xu B, Karayiorgou M . De novo synonymous mutations in regulatory elements contribute to the genetic etiology of autism and schizophrenia. Neuron 2016; 89: 940–947.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P et al. A method and server for predicting damaging missense mutations. Nat Methods 2010; 7: 248–249.
Acknowledgements
The current research was supported by research grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan; the Ministry of Health, Labour and Welfare of Japan; the Strategic Research Program for Brain Sciences from the Japan Agency for Medical Research and Development (AMED); the Brain Mapping by Integrated Neurotechnologies for Disease Studies (Brain/MINDS) project of AMED; Grant-in-Aid for Scientific Research on Innovative Areas ‘Glial assembly: a new regulatory machinery of brain function and disorders’ and Innovative Areas ‘Comprehensive Brain Science Network’.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Translational Psychiatry website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Kimura, H., Fujita, Y., Kawabata, T. et al. A novel rare variant R292H in RTN4R affects growth cone formation and possibly contributes to schizophrenia susceptibility. Transl Psychiatry 7, e1214 (2017). https://doi.org/10.1038/tp.2017.170
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tp.2017.170
This article is cited by
-
Proteomic insights into mental health status: plasma markers in young adults
Translational Psychiatry (2024)
-
Genetics and Schizophrenia
Current Behavioral Neuroscience Reports (2024)
-
The genetic architecture of schizophrenia: review of large-scale genetic studies
Journal of Human Genetics (2023)
-
Proteomic analysis of 92 circulating proteins and their effects in cardiometabolic diseases
Clinical Proteomics (2023)
-
The 22q11.2 region regulates presynaptic gene-products linked to schizophrenia
Nature Communications (2022)
