
Abstract
Elevated whole-blood serotonin and decreased plasma melatonin (a circadian synchronizer hormone that derives from serotonin) have been reported independently in patients with autism spectrum disorders (ASDs). Here, we explored, in parallel, serotonin, melatonin and the intermediate N-acetylserotonin (NAS) in a large cohort of patients with ASD and their relatives. We then investigated the clinical correlates of these biochemical parameters. Whole-blood serotonin, platelet NAS and plasma melatonin were assessed in 278 patients with ASD, their 506 first-degree relatives (129 unaffected siblings, 199 mothers and 178 fathers) and 416 sex- and age-matched controls. We confirmed the previously reported hyperserotonemia in ASD (40% (35–46%) of patients), as well as the deficit in melatonin (51% (45–57%)), taking as a threshold the 95th or 5th percentile of the control group, respectively. In addition, this study reveals an increase of NAS (47% (41–54%) of patients) in platelets, pointing to a disruption of the serotonin-NAS–melatonin pathway in ASD. Biochemical impairments were also observed in the first-degree relatives of patients. A score combining impairments of serotonin, NAS and melatonin distinguished between patients and controls with a sensitivity of 80% and a specificity of 85%. In patients the melatonin deficit was only significantly associated with insomnia. Impairments of melatonin synthesis in ASD may be linked with decreased 14-3-3 proteins. Although ASDs are highly heterogeneous, disruption of the serotonin-NAS–melatonin pathway is a very frequent trait in patients and may represent a useful biomarker for a large subgroup of individuals with ASD.
Similar content being viewed by others

Introduction
Autism spectrum disorders (ASDs) are complex, heterogeneous and multifactorial disorders characterized by impaired social communication and repetitive/stereotyped behaviors. The diagnosis of ASD currently relies entirely on patient clinical evaluation.1 Intensive investigations are underway to identify genetic, biochemical, electrophysiological or imaging markers that could contribute to screening and/or subgrouping diagnosis in a clinical setting. Large-scale genetic studies have pointed out a marked heterogeneity of ASD,2 and, at the present time, routine genetic testing only allows the etiological diagnosis of a minority of cases, although fast technological advances may gradually lead to enlargement of this group. Some imaging,3, 4 electrophysiological5 and eye-tracking6 studies have shown promising results as contributors to the diagnosis or to early screening tools, but they have not been translated into clinical practice so far.7
Among the different types of paraclinical examinations, biological markers are readily available, as well as are time and cost efficient. Several biological abnormalities have been reported in individuals with ASD, including neurochemical, immunological, endocrine or metabolic variations.8, 9, 10 Among these, elevated whole-blood serotonin (5-hydroxytryptamine) is the most replicated finding, reported in more than 25 studies.11, 12, 13 A deficit in melatonin (which derives from serotonin) has also been described in several studies on the basis of plasma or urine samples of individuals with ASD.14, 15, 16, 17, 18
Melatonin, a neurohormone mainly synthesized in the pineal gland during the night, is a biological signal of light/dark cycles and is considered as a major circadian synchronizer. It also displays antioxidant and neuroprotective properties and can directly modulate neuronal networks.19, 20 The phase and amplitude of the nocturnal melatonin peak display marked interindividual variations: although a consensus threshold of 10 ng l−1 in human plasma is classically used to determine the time of melatonin onset, in some individuals the nocturnal peak remains below this threshold.21 Pharmacological doses of melatonin (from 0.5 mg) can modulate circadian rhythms;22 thus it is often used in the treatment of sleep disorders, which are commonly reported in patients with ASD.23, 24, 25, 26, 27, 28
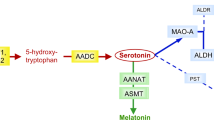
Conversion of serotonin into melatonin involves two sequential enzymatic steps (Figure 1a). The intermediate metabolite, N-acetylserotonin (NAS), displays intrinsic biological properties: it is an agonist of the TrkB receptor and may therefore share the neurotrophic properties of brain-derived neurotrophic factor, the canonical TrkB ligand.29, 30

Exploration of the serotonin-NAS–melatonin pathway in the blood. Two hundred and seventy-eight patients with ASD, their first-degree relatives (129 unaffected siblings and 377 parents) and 416 controls were sampled in the morning. Boxes indicate medians and quartiles. Groups were compared using the Wilcoxon two-sample test. (a) Overview of the serotonin-NAS–melatonin pathway. Serotonin is converted into NAS (N-acetylserotonin) by AANAT (arylalkylamine N-acetyltransferase, EC: 2.3.1.87), then converted into melatonin by ASMT (acetylserotonin N-methyltransferase, EC: 2.1.1.4). (b) Whole-blood serotonin (c) Platelet N-acetylserotonin (d) Plasma melatonin (1 nM=232 ng l−1). (e–g) Correlations between the three parameters in patients with ASD, tested as linear (e and f) or log-linear (g) correlations. (h) 6-Sulfatoxymelatonin measurements in overnight urine from a subgroup of 16 patients with ASD and 10 controls. ASD, autism spectrum disorder.
In a previous study,17 we reported alterations of peripheral serotonin and melatonin levels in patients with ASD. In the present research, we hypothesized that (i) the intermediate NAS might also be altered, (ii) alterations of the serotonin-NAS–melatonin pathway might constitute a possible biomarker for a subgroup of individuals with ASD and that (iii) they would be associated with specific clinical profiles. Thus, to address the issue of diagnostic validity and clinical correlates of impairments of the serotonin-NAS–melatonin pathway, our study was extended to a larger cohort of patients with ASD and their relatives, including the assessment of NAS as well as serotonin and melatonin.
Materials and methods
Ethics statement
The local Institutional Review Boards approved the study. Written informed consents were obtained after oral and written information from all adult participants of the study and from the children’s parents when subjects were under 18 years of age.
Subjects and clinical evaluations
Unrelated patients with ASD (n=278), their first-degree relatives (129 unaffected siblings and 377 parents, involving 171 trios, 28 index-mother pairs and 7 index-father pairs) and 416 sex- and age-matched controls were recruited into the Paris Autism Research International Sib-pair (PARIS) study at specialized centers in France. The patient characteristics and parameters are presented in Supplementary Table 1 and Supplementary Figure 2b. All individuals, including controls, were enrolled after a medical and psychiatric assessment using the Diagnostic Interview for Genetic Studies for adults and Kiddie-Schedule for Affective Disorders and Schizophrenia (K-SADS) for children. Probands with a total score >30 at the Social Responsiveness Scale (SRS)31 or for which a diagnosis of ASD was suspected, underwent additional screening with the Autism Diagnostic Interview-Revised with a parent32 and/or the Autism Diagnostic Observation Schedule.33 The final diagnosis of ASD was made when clinical assessment diagnosis coincided with algorithm-suggested diagnosis of autism on either or both of the Autism Diagnostic Interview-Revised and the Autism Diagnostic Observation Schedule. Autistic traits were measured using SRS in first-degree relatives of probands and in controls. Repetitive behaviors were assessed using the RBS (Repetitive Behavior Scale) for probands and their relatives.34 General medical history (including neurological, gastro-enterological and other conditions) was checked during interviews. For probands with ASD, a cognitive level was assigned using appropriate tests (Wechsler scales or Raven’s progressive matrices for nonverbal individuals). Intellectual disability was defined as verbal and performance IQ<70. Sleep difficulties were assessed in probands, relatives and controls by self-report (and/or parent questionnaire for probands who were themselves not sufficiently verbal to describe sleep behaviors) during interviews. Standard karyotyping, fragile X testing, MRI and electroencephalography were performed whenever possible. Individuals diagnosed with medical disorders related to ASD, such as fragile X syndrome, Rett syndrome or tuberous sclerosis, were excluded from the study.
Biochemical measurements
Individuals were asked to avoid food with a high content of tryptophan and/or serotonin (for example, chocolate, bananas) for 2 days preceding the blood draw. Individuals receiving exogenous melatonin or psychotropic drugs were excluded from the study. Blood samples were collected in the morning between 8:30 and 10:30 and fractioned, as previously published.17 Whole-blood serotonin was measured by high-performance liquid chromatography.35 Plasma melatonin was measured using a radioimmunoassay (RK-MEL, Bühlmann, Switzerland). NAS and 14-3-3 were determined in platelet pellets by radioenzymology36 and ELISA,37 respectively. Urine samples were collected overnight (2000–0800 hours) from 16 adult patients with high-functioning ASD and 10 adult controls. 6-Sulfatoxymelatonin was measured by a radioimmunological method as in Tordjman et al.16, 18
Statistical analyses
Statistical analyses and graphs were performed using JMP Pro 9 (SAS, Toronto, ON, Canada), R-statistical software (http://www.r-project.org/) and Stata 11.0 (StataCorp, College Station, TX, USA). Because the studied biochemical parameters were not normally distributed, categorical and nonparametric statistical tests were preferred. Error type I was defined as 0.05. ROC curves were analyzed and the areas under the curves compared according to the Hanley and McNeil method.38
Results
Disruption of the serotonin-NAS–melatonin pathway in patients with ASD and their relatives
Whole-blood serotonin, plasma melatonin and platelet NAS were assessed in individuals with ASD, their first-degree relatives and controls (Supplementary Table 1 for groups’ characteristics). To gain statistical power efficiency, the original serotonin and melatonin data collected for the present study were pooled with a smaller, independent set published in a previous report.17 Separate analysis indicated no significant differences between these two data sets (Supplementary Figure 1).
Individuals with ASD displayed elevated whole-blood serotonin (Figure 1b), as previously reported.8, 11, 12, 13 Analyses stratified by age (two groups: below and over 16 years old) were also performed, because serotonemia is known to be age-dependent.13, 39 On the basis of this stratification, hyperserotonemia remained significant for patients within the two age groups (Supplementary Figure 2a). Taking as a threshold the 95th percentile of the control group (830 nM for children <16 years old, 655 nM for individuals of 16 years or more), hyperserotonemia was observed in 40% of individuals with ASD. By contrast whole-blood serotonin was significantly elevated in ~15% of their first-degree relatives (17% for mothers, 14% for fathers and 14% for unaffected siblings—frequencies with 95% confidence interval and comparisons with controls and patients with ASD are summarized in Supplementary Table 2).
Plasma melatonin was significantly decreased in individuals with ASD and their relatives compared with controls (Figure 1d). Taking as a threshold the 5th percentile of the control group (0.07 nM or 16 ng l−1), melatonin deficit was observed in 51% of individuals with ASD, 26% of parents with a trend for a gender effect (31% of mothers and 21% of fathers, P=0.08) and 25% of unaffected siblings.
For the first time to our knowledge, the intermediate metabolite NAS, measured in blood platelets, was found to be significantly elevated in individuals with ASD and their relatives compared with controls (Figure 1c). Taking as a threshold the 95th percentile of the control group (44 nmoles/109 platelets), NAS was elevated in 47% of individuals with ASD, 19% of parents (24% of mothers and 15% of fathers, P=0.23) and 17% of unaffected siblings.
Platelet NAS was strongly correlated with plasma melatonin in patients with ASD (Figure 1g) and to a lesser extent in parents and controls (Supplementary Figure 3), suggesting common factors of deregulation. In contrast, a correlation between whole-blood serotonin and platelet NAS or plasma melatonin was observed neither in patients (Figures 1e and f) nor in controls (Supplementary Figure 3), pointing toward an independence of mechanisms leading to serotonin impairment on one hand, NAS and melatonin alterations on the other hand.
One limitation of our study is the assessment of melatonin only from plasma sampled in the morning, as melatonin displays marked nyctohemeral variations with a peak occurring at night.40 Thus, we extended melatonin assessment in a subgroup of 16 ASD patients and 10 controls by measuring 6-sulfatoxymelatonin (the main melatonin catabolite) in overnight urine. Consistent with previous studies,16, 18 nighttime 6-sulfatoxymelatonin excretion was significantly lower in patients than in controls, thus confirming a melatonin deficit in ASD (Figure 1h).
Serotonin, NAS and melatonin as biomarkers of ASD
Given that increased whole-blood serotonin and platelet NAS and decreased plasma melatonin are reported in approximately half of the patients with ASD in our study, we tested their accuracy to discriminate patients with ASD from controls. Figures 2a and b show that each potential biomarker performed well with areas under the curves >0.75. Platelet NAS was as powerful as blood serotonin in discriminating ASD patients from controls (P=0.33 for serotonin vs NAS). Plasma melatonin performed better than serotonin (P=0.02 for serotonin vs melatonin; Figure 2c).

Diagnostic performances of the serotonin-NAS–melatonin pathway parameters in ASD. (a) ROC curve analysis for serotonin, NAS, melatonin and the score combining all three as diagnostic markers of ASD. (b) AUC with confidence interval for each parameter. (c) Comparisons of AUC. (d) Design of the score combining impairments of serotonin, NAS and melatonin. The pathological thresholds were defined as the 95th percentile (for serotonin and NAS) or the 5th percentile (for melatonin) of the control group. (e) Diagnostic performances for ASD of the score. This score is based on n=230 controls and 229 patients, described for the whole population and after stratification by age category. ASD, autism spectrum disorder; AUC, area under the curve; NAS, N-acetylserotonin; ROC, receiver operating characteristic.
Considering that alterations of serotonin, NAS and melatonin are partially independent (Figures 1e–g), we tested whether a combination of these parameters could improve diagnostic performances. We designed a score taking into account serotonin, NAS and melatonin, with the 95th or 5th percentile of the control group as pathological thresholds (Figure 2d). Using this score, 80% sensitivity for 85% specificity was achieved for subjects displaying at least one abnormal parameter (score ⩾1), and the specificity reached 98.7% for subjects displaying two abnormal parameters or more (score ⩾2) with a 50% sensitivity; interestingly, the performance for this score was slightly better in children than in adults (Figure 2e).
Thus, the combination of serotonin, NAS and melatonin allows a good discrimination between ASD patients and controls.
Clinical correlates of the disruptions of the serotonin-NAS-melatonin pathway in ASD
We evaluated the association of serotonin, NAS and melatonin impairment with the clinical features of patients with ASD (Table 1). Overall, we did not observe a significant association between biochemical alterations and the severity of autistic symptoms or a comorbid intellectual disability. A trend was observed for an association between high blood serotonin in patients and lower social abilities (higher SRS score), but this trend was not consistent with the analysis of the ‘social’ (B) item of Autism Diagnostic Interview-Revised. Similarly, a nonsignificant trend was observed between low melatonin and higher scores on the repetitive behavior (D) item of Autism Diagnostic Interview-Revised, but this trend was not consistent with the analysis of the Repetitive Behavior Scale (RBS-R) scores. No association was observed between biochemical impairments and autistic traits (assessed by SRS and RBS-S) in first-degree relatives of patients (details not shown).
NAS might display neurotrophic properties as a TrkB agonist 29, 30 and early brain overgrowth was reported during infancy and the toddler years in ASD.41, 42 We then explored the associations between biochemical impairments and abnormal head sizes: ‘normal’ NAS tended to be associated with a lower frequency of small heads (Table 1).
Sleep abnormalities are frequently associated with ASD, and melatonin has a major role in synchronizing circadian rhythms. Thus, we assessed sleep difficulties in patients with ASD (Supplementary Figure 4) and explored the associations between alterations of serotonin, NAS and melatonin and sleep difficulties. Patients with melatonin deficit reported sleep-onset and sleep-maintenance insomnia significantly more frequently than patients with normal melatonin levels (Table 1). Conversely, patients reporting sleep abnormalities displayed significantly lower melatonin levels than patients without such difficulties (Supplementary Figure 5a). This association was not observed in first-degree relatives of patients.
In addition to self-reporting, we assessed sleep disorders by a Children’s Sleep Habit Questionnaire43 in a subgroup of ASD and control children, and by actigraphy and specific sleep questionnaires44, 45, 46 in a subgroup of high-functionning ASD and control adults (Supplementary Figure 4). The ASD patients displayed a high frequency of sleep disorders, including mostly insomnia (reported by 44% of patients) and delayed sleep onset. Children’s Sleep Habit Questionnaire analysis revealed no associations between sleep parameters and biochemical impairments in the subgroup of children with ASD (data not shown). In contrast, according to actigraphic recordings, elevated NAS was associated with lower diurnal motor activity (M10) and tended to be associated with lower amplitude between minimal and maximal activity (Supplementary Figure 5). A trend for an association between the melatonin deficit and the increased sleep latency was also observed. These results need further replication in larger groups. No association was observed between biochemical impairments and other comorbid psychiatric or somatic conditions.
Disruption of 14-3-3 proteins in platelets of patients with ASD
Melatonin synthesis involves two enzymes, AANAT and ASMT, known to form protein complexes with 14-3-3 scaffolding proteins.47 Interaction with 14-3-3 regulates this process by stabilizing and activating AANAT.48 In a previous study, we reported a decrease of ASMT activity in platelets of patients with ASD.17 Here, 14-3-3 was measured in platelets and found significantly decreased in patients with ASD (Figure 3). Thus, although obtained from peripheral tissues, these results suggest that the disruption of melatonin synthesis in patients with ASD may be linked with impairments of protein complex formation involving 14-3-3.

14-3-3 protein measurement in platelets. Forty patients with ASD and 70 controls were assessed. Boxes indicate medians and quartiles. Groups were compared using the Wilcoxon two-sample test. ASD, autism spectrum disorder.
Discussion
The exploration of the serotonin-NAS-melatonin pathway in our study confirmed the hyperserotonemia8, 11, 12, 13 and the melatonin deficit14, 15, 16, 17, 18 in ASD that has been reported in several studies, and it also revealed an increase of NAS in platelets, pointing towards a global disruption of this pathway in ASD.
Consistent with previous reports on serotonin and melatonin, impairments of the serotonin-NAS–melatonin pathway were frequent (40 to 51%) in this cohort of patients with ASD, and may thus be considered as possible biomarkers for a large subgroup of patients. In line with this idea, we found that combining the three parameters was sensitive and specific for discrimination of probands from controls.
The specificity of the combination of these biochemical biomarkers regarding other neurodevelopmental conditions should be further evaluated in future studies. However, previous reports49 and our preliminary data (Supplementary Figure 6) indicate that patients with intellectual disability not associated with autism (ID) or patients with attention-deficit hyperactivity disorder (ADHD) do not display as high blood serotonin levels as patients with ASD. To our knowledge, plasma melatonin has been documented neither in ID nor in ADHD patients. Our preliminary data suggest that ADHD or ID patients display a decreased level of plasma melatonin, but less pronounced than in ASD. ROC curve analysis indicated that whole-blood serotonin and plasma melatonin were discriminant between ADHD or ID patients and ASD patients, but not between ADHD or ID patients and controls (Supplementary Figure 6).
Disruption of the serotonin-NAS–melatonin pathway was also observed in first-degree relatives of patients with ASD, suggesting a genetic liability. This finding is consistent with previous reports of (i) increased serotonin among parents and siblings of patients with ASD,13, 50, 51, 52 (ii) decreased melatonin among parents17 and (iii) strong heritability of serotonin53 and melatonin54 reported in the general population. However, in our cohort, disruption of the serotonin-NAS–melatonin pathway does not always segregate in the families with ASD and rarely with a simple recessive/dominant model, suggesting a complex inheritance. Interestingly, mothers of patients with ASD would tend to display more pronounced biochemical impairments (elevated platelet NAS and low melatonin) than fathers. Considering that melatonin crosses the placental barrier, melatonin deficit in a mother might result in insufficient fetal exposure to melatonin in utero, and might thus be a prenatal, environmental susceptibility factor to ASD.
The search for clinical correlates of disruptions of the serotonin-NAS–melatonin pathway yielded no significant associations with ASD symptom severity or presence of intellectual disability. In addition, no association was found between biochemical impairments and diagnostic categories of ASD according to the DSM-IV (autism, Asperger syndrome, PDD-NOS—data not shown), which suggests that a defect in the serotonin-NAS–melatonin pathway is a shared feature across the spectrum.
Attempts to link whole-blood serotonin levels to clinical symptoms have been inconsistent. However, the overall evidence implicates the serotonin system in ASD: serotonin is crucial for embryonic brain development55, 56 and regulates the release of oxytocin whose signal alterations are important for ASD57 and which promotes social behavior in high-functioning ASD patients.58 Of note, oxytocin and serotonin interact in the human brain59 and their blood levels are negatively correlated in humans.60
One study reported an association between melatonin deficit and language difficulties in patients with ASD,16 but we were not able to replicate this finding. In our study, we observed a significant association between melatonin deficit and sleep problems in patients. Sleep difficulties are frequently associated with ASD and are a major concern for the patients and their families.61, 62 These results provide a rationale for the therapeutic use of melatonin in sleep disorders associated with ASD.23, 24, 25, 26, 27, 28, 63
A possible pitfall of this study is that melatonin levels were assessed only at one time point (over a 2-h window well after sleep offset) and not under dim light conditions. Obviously these conditions are not adequate for assessing the endogenous phase or amplitude of nocturnal melatonin secretion. However, they allow a good discrimination between patients and controls and are more convenient in a clinical setting than nocturnal melatonin assessment. Furthermore, nighttime 6-sulfatoxymelatonin excretion in a subset of patients confirmed a nocturnal melatonin deficit, consistent with previous studies.16, 18 To the best of our knowledge the circadian rhythm of platelet NAS has not been reported, whereas whole-blood or platelet serotonin only displays minor (<10%) circadian variations.64, 65
We reported for the first time an increase of the intermediate metabolite NAS in platelets of patients with ASD. Interestingly, NAS displays proper biological functions. At a metabolic level, it is an inhibitor of tetrahydrobiopterin synthesis,66 a cofactor of several pathways such as nitric oxide formation67 and tyrosine/bioamine synthesis. Thus, in addition to the possible consequences of alterations of serotonin and melatonin,6, 20 NAS accumulation may have implications for the pathophysiology of ASD.
The mechanisms of these impairments of the serotonin-NAS–melatonin pathway in ASD remain to be elucidated. Various studies have not succeeded in revealing a convincing mechanism for hyperserotonemia.68 Considering that melatonin synthesis represents only a small fraction (about 1%) of serotonin catabolism, it is unlikely that an impairment of melatonin synthesis would be sufficient to provoke a significant increase of serotonin. Our previous study17 suggested that melatonin deficit results from impaired synthesis as a consequence of a decrease in ASMT activity. This enzymatic alteration may be associated with genetic factors, including coding mutations in a subset of patients with ASD—although with a low frequency—and polymorphisms in the ASMT promoter (rs4446909 and rs5989681) with functional effects. The association finding was not fully replicated in an independent study, although a similar trend was observed.69 Interestingly, these SNPs have also been associated with bipolar disorder,70 depression71 and depressive symptoms in individuals with sleep phase delay.72 Here we show that 14-3-3 scaffolding protein levels are decreased in patients with ASD. As protein complex formation involving 14-3-3 regulate melatonin synthesis,47, 48 we propose that impairments of posttranslational regulations of AANAT and/or ASMT may participate in the mechanisms of the disruption of melatonin synthesis in ASD.
Although ASDs are considered as highly heterogeneous, this study reports disruptions of the serotonin-NAS–melatonin pathway as a highly sensitive and specific biomarker of ASD. Associations with sleep problems provide a rationale for melatonin therapy. Finally, these results open new hypotheses for the mechanisms of impairments of melatonin synthesis in ASD.
References
Lai MC, Lombardo MV, Baron-Cohen S . Autism. Lancet 2013; 383: 896–910.
Huguet G, Ey E, Bourgeron T . The genetic landscapes of autism spectrum disorders. Annu Rev Genom Hum Genet 2013; 14: 191–213.
Ecker C, Marquand A, Mourao-Miranda J, Johnston P, Daly EM, Brammer MJ et al. Describing the brain in autism in five dimensions—magnetic resonance imaging-assisted diagnosis of autism spectrum disorder using a multiparameter classification approach. J Neurosci 2010; 30: 10612–10623.
Koldewyn K, Yendiki A, Weigelt S, Gweon H, Julian J, Richardson H et al. Differences in the right inferior longitudinal fasciculus but not general disruption of white matter tracts in children with autism spectrum disorders. Proc Natl Acad Sci USA 2014; 111: 1981–1986.
Elsabbagh M, Mercure E, Hudry K, Chandler S, Pasco G, Charman T et al. Infant neural sensitivity to dynamic eye gaze is associated with later emerging autism. Curr Biol 2012; 22: 338–342.
Jones W, Klin A . Attention to eyes is present but in decline in 2-6-month-old infants later diagnosed with autism. Nature 2013; 504: 427–431.
Delorme R, Ey E, Toro R, Leboyer M, Gillberg C, Bourgeron T et al. Progress toward treatments for synaptic defects in autism. Nat Med 2013; 19: 685–694.
Lam KS, Aman MG, Arnold LE . Neurochemical correlates of autistic disorder: a review of the literature. Res Dev Disabil 2006; 27: 254–289.
Rossignol DA, Frye RE . A review of research trends in physiological abnormalities in autism spectrum disorders: immune dysregulation, inflammation, oxidative stress, mitochondrial dysfunction and environmental toxicant exposures. Mol Psychiatry 2012; 17: 389–401.
Ruggeri B, Sarkans U, Schumann G, Persico AM . Biomarkers in autism spectrum disorder: the old and the new. Psychopharmacology 2014; 231: 1201–1216.
Schain RJ, Freedman DX . Studies on 5-hydroxyindole metabolism in autistic and other mentally retarded children. J Pediatr 1961; 58: 315–320
Gabriele S, Sacco R, Persico AM . Blood serotonin levels in autism spectrum disorder: a systematic review and meta-analysis. Eur Neuropsychopharmacol 2014; 24: 919–929.
Leboyer M, Philippe A, Bouvard M, Guilloud-Bataille M, Bondoux D, Tabuteau F et al. Whole blood serotonin and plasma beta-endorphin in autistic probands and their first-degree relatives. Biol Psychiatry 1999; 45: 158–163.
Nir I, Meir D, Zilber N, Knobler H, Hadjez J, Lerner Y et al. Brief report: circadian melatonin, thyroid-stimulating hormone, prolactin, and cortisol levels in serum of young adults with autism. J Autism Dev Disord 1995; 25: 641–654.
Kulman G, Lissoni P, Rovelli F, Roselli MG, Brivio F, Sequeri P et al. Evidence of pineal endocrine hypofunction in autistic children. Neuroendocrinol Lett 2000; 21: 31–34.
Tordjman S, Anderson GM, Pichard N, Charbuy H, Touitou Y . Nocturnal excretion of 6-sulphatoxymelatonin in children and adolescents with autistic disorder. Biol Psychiatry 2005; 57: 134–138.
Melke J, Goubran Botros H, Chaste P, Betancur C, Nygren G, Anckarsater H et al. Abnormal melatonin synthesis in autism spectrum disorders. Mol Psychiatry 2008; 13: 90–98.
Tordjman S, Anderson GM, Bellissant E, Botbol M, Charbuy H, Camus F et al. Day and nighttime excretion of 6-sulphatoxymelatonin in adolescents and young adults with autistic disorder. Psychoneuroendocrinology 2012; 37: 1990–1997.
Claustrat B, Brun J, Chazot G . The basic physiology and pathophysiology of melatonin. Sleep Med Rev 2005; 9: 11–24.
Bourgeron T . The possible interplay of synaptic and clock genes in autism spectrum disorders. Cold Spring Harb Symp Quant Biol 2007; 72: 645–654.
Lewy AJ, Cutler NL, Sack RL . The endogenous melatonin profile as a marker for circadian phase position. J Biol Rhythms 1999; 14: 227–236.
Lewy AJ, Bauer VK, Hasler BP, Kendall AR, Pires ML, Sack RL et al. Capturing the circadian rhythms of free-running blind people with 0.5 mg melatonin. Brain Res 2001; 918: 96–100.
Wright B, Sims D, Smart S, Alwazeer A, Alderson-Day B, Allgar V et al. Melatonin versus placebo in children with autism spectrum conditions and severe sleep problems not amenable to behaviour management strategies: a randomised controlled crossover trial. J Autism Dev Disord 2011; 41: 175–184.
Andersen IM, Kaczmarska J, McGrew SG, Malow BA . Melatonin for insomnia in children with autism spectrum disorders. J Child Neurol 2008; 23: 482–485.
Malow B, Adkins KW, McGrew SG, Wang L, Goldman SE, Fawkes D et al. Melatonin for sleep in children with autism: a controlled trial examining dose, tolerability, and outcomes. J Autism Dev Disord 2012; 42: 1729–1737, author reply 1738.
Giannotti F, Cortesi F, Cerquiglini A, Bernabei P . An open-label study of controlled-release melatonin in treatment of sleep disorders in children with autism. J Autism Dev Disord 2006; 36: 741–752.
Cortesi F, Giannotti F, Sebastiani T, Panunzi S, Valente D . Controlled-release melatonin, singly and combined with cognitive behavioural therapy, for persistent insomnia in children with autism spectrum disorders: a randomized placebo-controlled trial. J Sleep Res 2012; 21: 700–709.
Paavonen EJ, Nieminen-von Wendt T, Vanhala R, Aronen ET, von Wendt L . Effectiveness of melatonin in the treatment of sleep disturbances in children with Asperger disorder. J Child Adolesc Psychopharmacol 2003; 13: 83–95.
Jang SW, Liu X, Pradoldej S, Tosini G, Chang Q, Iuvone PM et al. N-acetylserotonin activates TrkB receptor in a circadian rhythm. Proc Natl Acad Sci USA 2010; 107: 3876–3881.
Sompol P, Liu X, Baba K, Paul KN, Tosini G, Iuvone PM et al. N-acetylserotonin promotes hippocampal neuroprogenitor cell proliferation in sleep-deprived mice. Proc Natl Acad Sci USA 2011; 108: 8844–8849.
Constantino JN, Przybeck T, Friesen D, Todd RD . Reciprocal social behavior in children with and without pervasive developmental disorders. J Dev Behav Pediatr 2000; 21: 2–11.
Lord C, Rutter M, Le Couteur A . Autism Diagnostic Interview-Revised: a revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J Autism Dev Disord 1994; 24: 659–685.
Lord C, Risi S, Lambrecht L, Cook EH Jr, Leventhal BL, DiLavore PC et al. The autism diagnostic observation schedule-generic: a standard measure of social and communication deficits associated with the spectrum of autism. J Autism Dev Disord 2000; 30: 205–223.
Lam KS, Aman MG . The Repetitive Behavior Scale-Revised: independent validation in individuals with autism spectrum disorders. J Autism Dev Disord 2007; 37: 855–866.
Kema IP, Schellings AM, Hoppenbrouwers CJ, Rutgers HM, de Vries EG, Muskiet FA et al. High performance liquid chromatographic profiling of tryptophan and related indoles in body fluids and tissues of carcinoid patients. Clin Chim Acta 1993; 221: 143–158.
Brownstein M, Saavedra JM, Axelrod J . Control of pineal N-acetylserotonin by a beta adrenergic receptor. Mol Pharmacol 1973; 9: 605–611.
Kenney K, Brechtel C, Takahashi H, Kurohara K, Anderson P, Gibbs CJ Jr et al. An enzyme-linked immunosorbent assay to quantify 14-3-3 proteins in the cerebrospinal fluid of suspected Creutzfeldt-Jakob disease patients. Ann Neurol 2000; 48: 395–398.
Hanley JA, McNeil BJ . A method of comparing the areas under receiver operating characteristic curves derived from the same cases. Radiology 1983; 148: 839–843.
Ritvo E, Yuwiler A, Geller E, Plotkin S, Mason A, Saeger K et al. Maturational changes in blood serotonin levels and platelet counts. Biochem Med 1971; 5: 90–96.
Simonneaux V, Ribelayga C . Generation of the melatonin endocrine message in mammals: a review of the complex regulation of melatonin synthesis by norepinephrine, peptides, and other pineal transmitters. Pharmacol Rev 2003; 55: 325–395.
Courchesne E, Campbell K, Solso S . Brain growth across the life span in autism: age-specific changes in anatomical pathology. Brain Res 2011; 1380: 138–145.
Chaste P, Klei L, Sanders SJ, Murtha MT, Hus V, Lowe JK et al. Adjusting head circumference for covariates in autism: clinical correlates of a highly heritable continuous trait. Biol Psychiatry 2013; 74: 576–584.
Owens JA, Spirito A, McGuinn M . The Children’s Sleep Habits Questionnaire (CSHQ): psychometric properties of a survey instrument for school-aged children. Sleep 2000; 23: 1043–1051.
Buysse DJ, Reynolds CF, Monk TH, Berman SR, Kupfer DJ . The Pittsburgh Sleep Quality Index: a new instrument for psychiatric practice and research. Psychiatry Res 1989; 28: 193–213.
Horne JA, Ostberg O . A self-assessment questionnaire to determine morningness-eveningness in human circadian rhythms. Int J Chronobiol 1976; 4: 97–110.
Johns MW . A new method for measuring daytime sleepiness: the Epworth sleepiness scale. Sleep 1991; 14: 540–545.
Maronde E, Saade A, Ackermann K, Goubran-Botros H, Pagan C, Bux R et al. Dynamics in enzymatic protein complexes offer a novel principle for the regulation of melatonin synthesis in the human pineal gland. J Pineal Res 2011; 51: 145–155.
Obsil T, Ghirlando R, Klein DC, Ganguly S, Dyda F . Crystal structure of the 14-3-3zeta:serotonin N-acetyltransferase complex. a role for scaffolding in enzyme regulation. Cell 2001; 105: 257–267.
McBride PA, Anderson GM, Hertzig ME, Snow ME, Thompson SM, Khait VD et al. Effects of diagnosis, race, and puberty on platelet serotonin levels in autism and mental retardation. J Am Acad Child Adolesc Psychiatry 1998; 37: 767–776.
Kuperman S, Beeghly JH, Burns TL, Tsai LY . Serotonin relationships of autistic probands and their first-degree relatives. J Am Acad Child Psychiatry 1985; 24: 186–190.
Leventhal BL, Cook EH Jr, Morford M, Ravitz A, Freedman DX . Relationships of whole blood serotonin and plasma norepinephrine within families. J Autism Dev Disord 1990; 20: 499–511.
Cook EH Jr., Leventhal BL, Heller W, Metz J, Wainwright M, Freedman DX et al. Autistic children and their first-degree relatives: relationships between serotonin and norepinephrine levels and intelligence. J Neuropsychiat Clin Neurosci 1990; 2: 268–274.
Abney M, McPeek MS, Ober C . Broad and narrow heritabilities of quantitative traits in a founder population. Am J Hum Genet 2001; 68: 1302–1307.
Hallam KT, Olver JS, Chambers V, Begg DP, Mc Grath C, Norman TR et al. The heritability of melatonin secretion and sensitivity to bright nocturnal light in twins. Psychoneuroendocrinology 2006; 31: 867–875.
Coté F, Fligny C, Bayard E, Launay JM, Gershon MD, Mallet J et al. Maternal serotonin is crucial for murine embryonic development. Proc Natl Acad Sci USA 2007; 104: 329–334.
Bonnin A, Goeden N, Chen K, Wilson ML, King J, Shih JC et al. A transient placental source of serotonin for the fetal forebrain. Nature 2011; 472: 347–350.
Tyzio R, Nardou R, Ferrari DC, Tsintsadze T, Shahrokhi A, Eftekhari S et al. Oxytocin-mediated GABA inhibition during delivery attenuates autism pathogenesis in rodent offspring. Science 2014; 343: 675–679.
Andari E, Duhamel JR, Zalla T, Herbrecht E, Leboyer M, Sirigu A et al. Promoting social behavior with oxytocin in high-functioning autism spectrum disorders. Proc Natl Acad Sci USA 2010; 107: 4389–4394.
Mottolese R, Redouté J, Costes N, Le Bars D, Sirigu A . Switching brain serotonin with oxytocin. Proc Natl Acad Sci USA 2014; 111: 8637–8642.
Hammock E, Veenstra-van der Weele J, Yan Z, Kerr TM, Morris M, Anderson GM et al. Examining autism spectrum disorders by biomarkers: example from the oxytocin and serotonin systems. J Am Acad Child Adolesc Psychiatry 2012; 51: 712–721.
Richdale AL, Schreck KA . Sleep problems in autism spectrum disorders: prevalence, nature, and possible biopsychosocial aetiologies. Sleep Med Rev 2009; 13: 403–411.
Cortesi F, Giannotti F, Ivanenko A, Johnson K . Sleep in children with autistic spectrum disorder. Sleep Med 2010; 11: 659–664.
Rossignol DA, Frye RE . Melatonin in autism spectrum disorders: a systematic review and meta-analysis. Dev Med Child Neurol 2011; 53: 783–792.
Wirz-Justice A, Schellings MJ, Feer H . Diurnal and seasonal variations in human platelet serotonin in man. J Neural Transm 1977; 41: 7–15.
Malyszko J, Urano T, Knofler R, Taminato A, Yoshimi T, Takada Y et al. Daily variations of platelet aggregation in relation to blood and plasma serotonin indiabetes. Thromb Res 1994; 75: 569–576.
Psarakis S, Brown GM, Grota LJ . Analgesia induced by N-acetylserotonin in the central nervous system. Life Sci 1988; 42: 1109–1116.
Klemm P, Hecker M, Stockhausen H, Wu CC, Thiemermann C . Inhibition by N-acetyl-5-hydroxytryptamine of nitric oxide synthase expression in cultured cells and in the anaesthetized rat. Br J Pharmacol 1995; 115: 1175–1181.
Pagan C, Delorme R, Launay J-M, Bourgeron T . Alterations of the serotonin–melatonin pathway in autism spectrum disorders: biological evidence and clinical consequences. In: Powell CM, Monteggia LM (eds). The Autisms—Molecules to Model Systems. Oxford University Press: New York, NY, USA, 2013; 240–273
Toma C, Rossi M, Sousa I, Blasi F, Bacchelli E, Alen R et al. Is ASMT a susceptibility gene for autism spectrum disorders? A replication study in European populations. Mol Psychiatry 2007; 12: 977–979.
Etain B, Dumaine A, Bellivier F, Pagan C, Francelle L, Goubran-Botros H et al. Genetic and functional abnormalities of the melatonin biosynthesis pathway in patients with bipolar disorder. Hum Mol Genet 2012; 21: 4030–4037.
Galecki P, Szemraj J, Bartosz G, Bienkiewicz M, Galecka E, Florkowski A et al. Single-nucleotide polymorphisms and mRNA expression for melatonin synthesis rate-limiting enzyme in recurrent depressive disorder. J Pineal Res 2010; 48: 311–317.
Kripke DF, Nievergelt CM, Tranah GJ, Murray SS, McCarthy MJ, Rex KM et al. Polymorphisms in melatonin synthesis pathways: possible influences on depression. J Circadian Rhythms 2011; 9: 8.
Acknowledgements
We thank the patients and their families, and the controls who accepted to participate in this study. The Clinical Investigation Centers of Robert-Debré and Henri Mondor Hospitals obtained and processed blood samples, the Hematology departments from both hospitals (Dr MF Hurtaud and Professor M Imbert) performed platelet counts. Dr E Gayat provided help with statistics. This work was supported by the Institut Pasteur, CNRS, INSERM, AP-HP, University Paris Diderot, the Bettencourt-Schueller foundation, the Orange foundation, the FondaMental foundation, the Conny-Maeva foundation, the Cognacq-Jay foundation, the ANR (SynDivAutism) and the Neuron-ERANET (EUHF-AUTISM).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Translational Psychiatry website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Pagan, C., Delorme, R., Callebert, J. et al. The serotonin-N-acetylserotonin–melatonin pathway as a biomarker for autism spectrum disorders. Transl Psychiatry 4, e479 (2014). https://doi.org/10.1038/tp.2014.120
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tp.2014.120
This article is cited by
-
Analysis of salivary steroid hormones in boys with autism spectrum disorder
BMC Psychiatry (2023)
-
Impact of IDO activation and alterations in the kynurenine pathway on hyperserotonemia, NAD+ production, and AhR activation in autism spectrum disorder
Translational Psychiatry (2023)
-
Glutamine increases stability of TPH1 mRNA via p38 mitogen-activated kinase in mouse mastocytoma cells
Molecular Biology Reports (2023)
-
Biological correlates of altered circadian rhythms, autonomic functions and sleep problems in autism spectrum disorder
Annals of General Psychiatry (2022)
-
Association between cord blood metabolites in tryptophan pathway and childhood risk of autism spectrum disorder and attention-deficit hyperactivity disorder
Translational Psychiatry (2022)
