
Abstract
Vernalization is required for floral initiation in Dendrobium. Interestingly, those beneficial effects can also be achieved by exogenous cytokinin application in greenhouses. Thus, an as yet unknown crosstalk/interaction may exist between vernalization and cytokinin signaling pathways. In this study, we showed, by de novo transcriptome assembly using RNA-seq data from both vegetative and reproductive tissue samples, that some floral transition-related genes—DnVRN1, FT, SOC1, LFY and AP1—were differentially expressed in low-temperature-challenged (LT) or thidiazuron (TDZ)-treated plants, compared to those mock-treated (CK). Both LT and TDZ upregulated SOC1, LFY and AP1, while the upregulation of DnVRN1 and FT was only LT-induced. We further found that LT promoted the upregulation of some key cytokinin signaling regulators, including several cytokinin biosynthesis-related genes and type-B response regulator (RR)-encoding genes, and that both LT and TDZ triggered the significant upregulation of some marker genes in the gibberellin (GA) signaling pathway, indicating an important low temperature-cytokinin-GA axis in flowering. Our data thus have revealed a cytokinin-GA signal network underlying vernalization, providing a novel insight into further investigation of the molecular mechanism of floral initiation in Dendrobium.
Similar content being viewed by others

Introduction
Vernalization is the process of promoting flowering in certain plants by prolonged exposure to the cold of winters. In Arabidopsis, vernalization represses the expression of FLOWERING LOCUS C (FLC), a potent flowering repressor, and this suppression requires VERNALIZATION INSENSITIVE 3 (VIN3)1, VERNALIZATION 2 (VRN2)2, and VERNALIZATION 1 (VRN1)3. As a MADS-box transcriptional regulator, FLC inhibits floral initiation by reducing the expression of flowering-time integrators including FLOWERING LOCUS T (FT) and SUPPRESSOR OF OVEREXPRESSION OF CONSTANS 1(SOC1). Vernalization-induced FT and SOC1 then activate floral meristem-identity genes such as LEAFY (LFY) and APETALA 1 (AP1), eventually leading to floral initiation.
Cytokinins, an important class of phytohormones, have long been considered to play a role in floral transition4. Two recent reports have demonstrated that endogenous cytokinin level is significantly increased in vernalization-induced reproductive development of Brassica napus plants5, and that exogenous application of N6-benzylaminopurine (BAP), a synthetic cytokinin, promotes flowering in Arabidopsis grown in non-inductive short days through transcriptional activation of the FT paralogue TSF and SOC16. In addition, by transcriptomic analysis cytokinins have been found to interplay with gibberellins (GA) in producing systemic flowering signal(s) in leaves of Glycine max7.
Dendrobium is one of the largest genera of the Orchidaceae and consists of more than 1200 species8, most of which are important commercial orchids that account for 20% of total orchid sales. They fall into two categories, nobile-type and phalaenopsis-type, which in horticulture are usually sold as potted and cut flowers, respectively9. The potted nobile-type Dendrobium requires moderate low temperatures (vernalization) for flower induction, except for some cultivars that are regulated by photoperiod10,11. While cold treatment is effective, its effects are of regional variations. To meet market demand, a growing interest has been shown in the control of flowering time. Cytokinin treatment was demonstrated to stimulate flowering as early as 1978 in a sympodial orchid hybrid Dendrobium Louisae12, but the underlying molecular mechanisms remain largely unknown.
We recently developed an effective method of flowering time control in nobile-type Dendrobium using thidiazuron (TDZ), a potent synthetic cytokinin-like compound, by which plants flowered 10~15 days earlier than cold-treated plants, even in the absence of cold in greenhouses13. Based on this fact, we deduce that an unknown crosstalk/interaction may exist between vernalization and cytokinin signaling pathways sustaining the floral initiation in D. nobile. To explore this underlying molecular mechanism, in this study differential gene expression profiles were investigated in the transcriptomic scope of the wild-type, cold- and TDZ-treated plants. This transcriptomic analysis was based on the de novo transcriptome assembly platform, Trinity14, using RNA-seq data from both vegetative (pseudobulb, leaf and root) and reproductive (axillary bud) tissue samples. Our data suggest an important low temperature-cytokinin-GA axis regulating flowering time of D. nobile.
Results
De novo assemblies of the transcriptomes of D. nobile plants
Two-year old adult plants were randomly selected and exposed to 4 °C (LT-group), given one-time spray of 20 mg L−1 of TDZ (TDZ-group), or left untreated (control group) for 30 days. Leaves, roots, pseudobulbs, and axillary buds of each group were collected and pooled at the beginning and every five days of this duration. Total RNA was then extracted for RNA-seq using an IlluminaHiSeq™ 2000 platform. The subsequent de novo transcriptome assemblies using the Trinity platform14, which was based on the 110 M raw reads obtained from the RNA-seq, thus yielded 99,686 unigenes, including 38,309 clusters and 61,377 singletons (Table 1). The average unigene size was 684 bp, and 21% of the unigenes were greater than 1000 bp in length (Figure S1).
Functional annotation and classification of D. nobile Unigenes
Unigene annotation were performed by BlastX against the protein databases NR, Swiss-Prot, KEGG and COG (E-value < 1e-5). The annotated unigenes accounted for 53.9% of all the assembled unigenes. Of these 53,685 annotated unigenes, 20,751 fell into 25 COG functional categories (Figure S2) including the “signal transduction mechanisms” (13.4%), and 37,784 were annotated to 53 GO terms (Figure S3) including the “reproduction” (11.3%) and “signaling” (8.2%). By that GO enrichment analysis, we also noticed that some unigenes were associated with nucleic acid binding transcription factor activity (2%), representing previously reported chromatin modifications in floral transition15. To further identify the biological pathways in which the annotated unigenes get involved, 26,624 annotated unigenes were mapped to 125 reference canonical pathways in the KEGG pathway database (Table S2), among which 1,305 unigenes (4.33%) were assigned to “plant hormone signal transduction” and 336 unigenes (1.11%) were grouped as “zeatin biosynthesis”.
Homologs in relation to floral transition
Based on the above functional prediction and enrichment analysis, we identified some D. nobile homologs of Arabidopsis FT, SOC1, LFY and terminal flower 1 (TFL1)16, which are the key flowering-time integrator genes (Table S3). FLC in Arabidopsis, and VRN1, VRN2, and VRN3 (cereal FT) in temperate cereals17,18 are well known as critical vernalization-induced flowering regulators. However, as shown in a recent report19, FLC and VRN2 homologs were not found in our dataset, but the D. nobile VRN1 (DsVRN1) (unigene ID: CL16305.Contig1_TRA) identified, supporting the difference demonstrated in flowering control between dicot and monocot plants18. Many genes related to chromatin regulation were also identified, which showed that, like the vernalization pathway in Arabidopsis, cold and cytokinin may use chromatin regulators to control flowering time in D. nobile. There were 32 unigenes annotated as genes encoding zinc finger protein CONSTANS (Table S4), the first B-box protein identified in Arabidopsis playing a central role in the photoperiodic flowering pathway20.
Additionally, 64 unigenes encoding MADS-box transcription factors were identified, including the homologs of important flowering time genes such as SVP, SOC1 and AP1/VRN1 (cereals) (Table S5). Of these unigenes, 26 had a conserved MADS-box domain, and 24 of them had a K-box domain following the MADS-box domain. The phylogenic tree constructed based on the MADS-box domain is shown in Fig. 1. The 26 putative MADS-box transcription factors were mainly classified into AP1/FUL, AGL20-like, E-, C/D-, and SVP clades, while FLC-like genes were absent in the current transcriptomic analysis.

Evolutionary analyses based on conserved MADS-box domains were conducted in MEGA5 using the maximum likelihood method. The bootstrap consensus tree inferred from 300 replicates is taken to represent the evolutionary history of the taxa analyzed. The analysis involved 26 MADS-box domain containing genes identified in this study and 22 MADS-box genes from Arabidopsis under gene bank accession number (GI) as shown in Table S9.
Homologs in relation to cytokinin metabolism and signaling
The enzymes encoded by IPT, CYP and LOG catalyze the rate-limiting step in cytokinin biosynthesis, and CKX encodes the key enzyme for cytokinin metabolism21. Homologs of IPT, CYP and LOG were all identified in the D. nobile transcriptome (Table 2), and there were seven unigenes annotated as CKX. Response regulators (RRs) are the downstream nuclear-localized components of the two-component system that are believed to be the major regulators of cytokinin response in plants22. There are 22 RRs in Arabidopsis, which were originally divided into two large classes (type-A and type-B) based on protein sequences, domain structures and transcriptional induction by cytokinin. Type-B RRs appear to act as the positive regulators of cytokinin signaling, while type-A RRs act as the negative regulators, forming a negative feedback loop. The homologs of both type-A and type-B RRs were identified in our dataset, and four of them were validated by real-time PCR (Table 2). All of these cytokinin-associated genes would helpful for us to investigate the mechanism of cytokinin induced floral initiation in D. nobile.
Differentially expressed genes in cold-treated plants
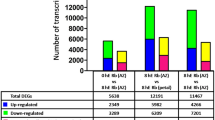
Total RNAs were extracted from leaves, pseudobulbs, axillary buds and roots collected and then mixed in a mass ratio of 1:1:1:1. Mixed RNAs from the control, cold-treated, and TDZ-treated D. nobile plants were used to construct three DGE libraries and then sequenced. The sequencing quality evaluation and alignment statistics are shown in Table S6. The percentage of all low quality reads containing only adaptors or N reads was less than 2%. After filtering the low quality tags, the total numbers of clean tags in the control, cold-treated, and TDZ-treated libraries were 12.23, 11.73 and 11.93 million, respectively. The number of tags that could be mapped to the transcriptomic sequences amounted to 10.17 (83.15%), 9.79 (83.52%), and 10.11 (84.79%) million respectively. The number of clean ta for each gene was calculated, and the genes that were differentially expressed were identified according to the methods described by Audic and Claverie23.
A total of 2,361 differentially expressed genes, including 1,060 up-regulated and 1,301 down-regulated genes, were identified in the cold-treated materials. The expression of nine genes, including a membrane-associated type I inositol 1,4,5-trisphosphate 5-phosphatase encoding gene (CL10301.Contig2_TRA) and a cell-wall structure protein encoding gene (CL3912.Contig1_TRA), was completely inhibited by cold, while a total of 40 genes were activated (Table S7). Based on GO functional classification (Figure S4), most of the gene sets regulated by cold were mainly correlated to cellular and organelle component organization.
Cluster analysis of flowering-time associated genes was conducted. Two homologs of FT, Unigene 8516 and Unigene 47953, were regulated by cold. Unigene 8516, annotated as DnTSF6 by sequence blast, was inhibited by cold, whilst unigene 47953, a gene sharing high homology with DnFT identified by Liang et al.19, was obviously up-regulated by cold. Transcription of the homolog of SOC1, another floral integrator, was apparently activated by cold, too (Unigene44321, Fig. 2). The results showed that, the expression of floral identity genes LFY and AP1 was up-regulated, while that of upstream repressor MSI1 and CLF16 was down-regulated (Fig. 2). Real-time PCR using axillary buds showed the expression of AP1 and LFY peaked at 20 d post-vernalization (Fig. 3), and hit the bottom at 5 d after the completion time of floral transition reported by Liang et al.19. As noted previously, there were no homologs of FLC identified in this study. The unigene CL16053.contig2_TRA, a homolog of Arabidopsis AP1, was identified as DnVRN1, the cereal VRN1 homolog in D. nobile. Its expression was significantly up-regulated by cold. Another 12 transcripts with homology to AP1 were also regulated by cold, and the expression of the homologs of AG, AGL16, AGL21, SVP and ZMM17 were regulated by cold as well (Table 3). All of these MADS-box genes were candidates for further functional analysis to clarify the underlying mechanism of cold-induced floral initiation. Besides, CLF, ULT1 and REF6 are important members in chromatin methylation of FLC and VRN1, their homologs were also significantly regulated by cold in D. nobile.

Each column represents an experimental sample (CK-control, LT-cold treatment, and TDZ-TDZ treatment), and each row represents a gene, the annotation of which were listed on the left. Expression differences were determined by RPKM (Reads Per kb per Million reads) values calculated according to the method provided by Mortazavi and Williams (2008), and shown in different colors. Red indicates high expression, and green indicates low expression.

The relative quantity of gene expression in axillary buds of control, cold-treated and TDZ-treated plants were calculated compared with the control gene DnActin. And represented by CK, LT, and TDZ, respectively, which showed in the figure marked by different color. Error bars indicate the range of variation in three biological repeats.
Results above showed a somewhat conserved molecular mechanism of vernalization in D. nobile. Considering the objective of this study, we analyzed the expression profiles of the cytokinin-associated genes. The homologues of IPT and CYP735A1, which encode the two key enzymes for cytokinin biosynthesis, were activated by cold (Table S8). Expression levels of type-B RRs in cold-treated plants increased, while those of type-A RRs were down-regulated (Table S8, Fig. 4), showing an improved cytokinin signal transduction.

Each column represents an experimental sample (CK-control, LT-cold treatment, and TDZ-TDZ treatment), and each row represents a gene. 54 DEGs that were significantly regulated (p-value < 0.05) were analyzed in pairwise comparisons (CK-VS-LT, CK-VS-TDZ,and LT-VS-TDZ). Expression levels were determined by RPKM (Reads Per kb per Million reads) values calculated from Illumina read counts, according to the method provided by Mortazavi and Williams (2008). Expression differences are shown in different colors. Red indicates high expression, and green indicates low expression.
Interes, we found that the transcription of six genes which shared homology with the cereal transcription factor GAMyb (GAM1) was significantly up-regulated by cold (Fig. 2). GAM1 is an early GA response gene encoding a key molecule in synthesizing and secreting hydrolytic enzymes in cereal aleurone24. The results indicated the nutrition metabolism regulated by a cold-induced GA signal. But what was contradictory to this was that the down-regulated expression of the GA receptor GID1 encoding genes, and the activation of DELLA protein GAI encoding genes in the cold-treated plants (Fig. 2).
The roles of cytokinin and GA in cold-induced flowering were further revealed by real-time PCR, using RNAs extracted from axillary buds. The GA20ox homolog (CL2388.Contig3) was immediately up-regulated by cold, and the transcription of the GA3ox homolog (Unigene24590_TRA) obviously increased at 4 d. The results indicated a cold-regulated GA signaling, along with the fact that the transcription of the GA-regulated protein encoding cDNA (CL5968.Contig1_TRA) increased at 3d and 4d post-vernalization. The transcription of GA20ox homologue decreased from 2d after treatment and hit the bottom at 5d, and the GA3ox homologue was inhibited at the same time (Fig. 5). In addition, the relative transcriptional level of Unigene494_TRA obviously increased at 5d, 30d and peaked at 20d after cold-treatment (Fig. 3). This gene was annotated as the homolog of AtLOG8, which encodes the key enzyme of the last step of cytokinin activation in Arabidopsis. In accordance with this, an expression peak of two cytokinin trans-hydroxylase encoding genes, Unigene39396_TRA and CL7022.Contig1_TRA, was observed at 5 d (Fig. 5), which showed an up-regulation of cytokinin biosynthesis induced by cold during this time. Besides, another obvious activation of GA20ox and GA3ox homologues appeared at 9 d and 11 d, respectively. These changes together with the inhibition of GA2ox homologue (Unigene3219_TRA) at 14d indicated an activation of GA signaling during the late stage of the cold-induced floral transition.

The relative quantity of gene expression in axillary buds of control (CK), cold-treated (LT) and TDZ-treated (TDZ) plants were calculated compared with the control gene DnActin. Error bars indicate the range of variation in three biological repeats.
Differentially expressed genes in TDZ-treated plants
The analysis of DGE sequencing resulted in 588 up-regulated genes and 693 down-regulated genes in the TDZ group compared to the control group, which was half as many as the genes regulated by cold. The nine genes completely inhibited by cold were also inhibited by TDZ. Five genes including a starch phosphorylase encoding gene (Unigene13605_TRA) was activated by TDZ (Table S7), and the transcription of the two TPS1 (Trehalose-6-P synthase1) homologues (Unigene57157_TRA, CL3080.Contig2_TRA) were up-regulated (Fig. 2). It was reported that TPS1 was regulated by sucrose, and activated the expression of FT in leaves through the miR156 pathway25. The activation of a starch phosphorylase encoding gene and TPS1 homologues indicated the nutrition metabolism involved in TDZ-induced flowering.
Homologues of the floral integrator SOC1 were up-regulated in TDZ-treated plants, but apparent changes of CL16053.contig2_TRA (DnVRN1) were not observed (Table 3). The floral meristem identity genes LFY and AP1 were activated (Fig. 2, Table 3), indicating the complete floral transition by TDZ. Results of the real-time PCR analysis showed that the transcription of AP1 and LFY peaked at 20 d after TDZ treatment, as in the cold-treated axillary buds, and their transcriptional level were much higher than that in the cold-treated plants. Consistent with these real-time PCR results, the axillary buds of the TDZ-treated plants were observed to swell and bulge out about 1–2 cm (Fig. 6). Unlike the subsequent flower development of the cold treated plants occurring under warmer conditions, the TDZ-induced floral initiation was followed by inflorescence and floral organ development, consistent with the observation of the increased transcription of ERECTA encoding cDNAs (Fig. 4). The latter play important roles in meristem differentiation and floral organ development.

Pictures of control plants (CK), cold-treated (LT) and TDZ-treated plants before treatment (0 d) and at 30 d after treatment were showed, no apparent morphology changes of axillary buds observed in control and cold-treated plants. But an obvious development of inflorescence appeared in TDZ-treated plants. And the close-up observance of axillary buds at 0, 5, 10, 15, 20, 25, and 30 d after TDZ-treatment showed that the buds bulged at 15 d, the bottom panel.
There was no surprise to observe the feedback inhibition of the endogenous cytokinin level in the TDZ treated D. nobile, which was supported by the up-regulation of cytokinin dehydrogenase (CKX) encoding genes. CKX is a key enzyme functioning in cytokinin catabolism. Besides, type-A RRs which negatively regulate cytokinin signaling were significantly up-regulated by TDZ, while the positive regulators type-B RRs were down-regulated (Fig. 4, Table S8). The expression of a CKX homologue, Unigene45836_TRA, increased significantly at 2 d post-application of TDZ, and maintained at a low level in a few days, then increased at 11 d again. These changes were consistent with the upregulation of CL12022.Contig1_TRA, a type-A RR encoding cDNA (Fig. 5), indicating the decreased endogenous cytokinin biosynthesis.
We also noticed that the transcription of GA receptor gene GDI1 was up-regulated by TDZ, and the DELLA protein GAI and RGL encoding genes were down-regulated (Figs 3 and 4), suggesting the enhanced GA signaling induced by TDZ. Indeed, the GA-regulated protein encoding cDNA, CL5968.Contig1_TRA, was significantly up-regulated by TDZ at 1d, along with the transcriptional activation of the GA20ox homologue at 7d and the CL5986.Contig1_TRA at 8d (Fig. 5).
Discussion
D. nobile is one of the most widespread ornamental members of orchid family, but the control of flowering time still remains a bottleneck in production. Our data in this study has demonstrated that the exogenous application of TDZ may effectively induce flowering in D. nobile grown in greenhouses, thus providing a cost-effective method for floral transition in horticulture. Cytokinins were proved to be effective in inducing flowering in some plants, but there are few reports on the crosstalk between vernalization and cytokinin signaling pathways. The molecular mechanism sustaining the TDZ-induced floral transition needs to be clarified.
There are very limited genetic information of D. nobile, except for the recently reported expressed sequence tags (ESTs) from flowering buds of Orchidaceae species26 and the genomic sequences of Phalaenopsis equestris27 and Dendrobium officinale28. The cDNA data acquired in this study was much more than that acquired from the transcriptomes of other orchids, i.e. oncidium29, cymbidium30, and Phalaenopsis31. All the flower development-related genes identified were valuable for further investigation of the floral transition in D. nobile. Starting at this point, we studied the molecular mechanism underlying the crosstalk between vernalization and TDZ-induced floral initiation by comparative transcriptomic analysis.
The up-regulation of DnVRN1, floral integrator FT and SOC1, and flower identity genes AP1 and LFY by cold, together with the differential expression of CLF, REF6 and many chromatin regulation associated genes, showed a somewhat conserved molecular mechanism of flowering used by D. nobile. Unexpectedly, except for the up-regulation of AP1 and LFY, we find little flowering-time associated genes similarly regulated by cold and TDZ. But the membrane-associated type I inositol 1,4,5-trisphosphate 5-phosphatase encoding gene (CL10301.Contig2_TRA), and the cell-wall structure protein encoding gene (CL3912.Contig1_TRA) were inhibited by both cold and TDZ. These two genes are both regulators of cell wall synthesis. These results indicated that cell wall reorganization was involved in floral initiation induced by cold or TDZ in D. nobile, as reported in Arabidopsis32. An extensive overlap between cytokinin and glucose-signaling pathways was proposed by Kushwah and Laxmi33, and the regulation of TPS1 by TDZ in this study suggested a role of sugar signaling in flowering of D. nobile. Our data also support the roles of sugar signaling in cold-induced floral initiation by exhibiting the cold-induced upregulation of a GA-response gene GAM1, which encodes a key molecule involved in synthesizing and secreting hydrolytic enzymes in cereal aleurone24.
By the comparative transcriptomic analysis shown in this study, we demonstrated that GA signaling was significantly improved during both vernalization and TDZ induced floral transitions. GA was proved to play important roles in floral initiation but not in flower development, and many reports have pointed out that vernalization makes plants more sensitive to GA34. Recently, it has been revealed that DELLA proteins, the repressors of GA signaling, interact with FLC in vernalization-induced flowering35. The positive effects of GA on the termination of vegetative development have been reported in Arabidopsis36, but still remain unexplored in D. nobile. According to the transcriptomic analysis presented in this study, a crosstalk between GA and cytokinin contributes to vernalization-induced floral initiation. The temporal transcriptional activation of the GA-response genes appeared immediately after the cold treatment, and in a latter stage, 7–9 d post-vernalization. In these two stages, cytokinin signaling was also enhanced, which was supporting by the significant up-regulation of cytokinin synthase encoding gene (Unigene39396_TRA, CL7022.Contig1_TRA, Fig. 3), and the transcriptional changes of type-A RR and type-B RR genes. Consistent with these data from the cold-treated plants, an enhanced GA signaling was also observed in the TDZ group with the fact that the TDZ application induced a ten-fold increase in the expression levels of CKX, GA3ox and GA20ox at 2 d, the peak expression of GA3ox and GA20ox at 7d (Fig. 5), together with the up-regulation of GID1 and down-regulation of DELLA protein GAI and RGL encoding genes (Figs 2 and 4). Interestingly, the expression of Unigene45836_TRA (CKX) was also significantly increased in a latter stage, indicating the sustaining cytokinin signaling activation in the late stage of the duration of TDZ treatment (Fig. 5).
Morphology analysis of the axillary buds showed that swelling appeared at 10d after TDZ application, while no changes were observed in the LT-treated plants (Fig. 6). In Glycine max, by transcriptomic analysis it was indicated that cytokinins could promote flowering by cytokinin signaling itself through B-type RRs, or play negative roles via inhibiting GA, which may activate the flowering integrator SOC137. AP1 may suppress the biosynthesis and activate the degradation of cytokinin in the establishment of determinate floral meristems in Arabidopsis38, which indicates that cytokinins function early in floral transition. According to the results of this study, it can be deduced that the rapid accumulation of cytokinins in meristems can be induced by the application of TDZ, facilitating the positive effects of cytokinins imposed on floral initiation. Meanwhile, the feedback inhibition of endogenous cytokinin level may result in the enhancement of GA signaling, which was important for further flower development. For the vernalized plants, the enhanced GA signaling induced by cold was followed by the activation of cytokinin biosynthesis genes at 5d (Fig. 5), leading to the floral initiation similar to that in the TDZ treated plants. The inhibitory effects of GA on cytokinin during vernalization may cause a delay of flowering compared to the TDZ-treated plants. More details and the molecular mechanism of the subsequent flower development need to be further investigated.
Methods
Plant materials, growth conditions, cold- and TDZ-treatment
Plants of D. nobile were grown without regulation of photoperiod in a greenhouse at the College of Life Sciences at South China Agricultural University (China). Two-year old adult plants were selected on Oct. 9, 2012, and 200 of them were moved to an air-conditioned storage room with a temperature setting of 4°Cduring the night (7:00 p.m. to 7:00 a.m.) for 30 d. Another 400 plants were divided into two groups, one of which was sprayed with 20 mg L−1 of TDZ. All of the plants were watered every week. Organs (i.e., leaf, pseudobulb, root, and axillary buds) from cold-treated, TDZ-treated, and control plants were sampled at 0 d, 5 d, 10 d, 15 d, 20 d, 25 d, and 30 d after treatment. The number of plants sampled was 20 every time for every treatment. All samples were frozen in liquid nitrogen and stored at −80 °C.
RNA extraction, library preparation, and Illumina sequencing
Total RNA was extracted from sampled tissues using a Qiagen RNeasy Kit according to the manufacturer’s instructions. Total RNA extracted from four organs (i.e., leaf, pseudobulb, root, and axillary buds) at different time from one treatment were mixed at an equal mass ratio, and then acquired 3 RNA samples from cold-treated, TDZ-treated and control plants, respectively. For transcriptome sequencing, 3 samples were mixed at a mass ratio of 1:1:1, then mixed total RNA was used for messenger RNA isolation with polyA selection and subsequent library construction using the TruSeq RNA sample preparation protocol from Illumina (San Diego, CA). Two biological replicates were sequenced and analysed for each of the nine tissue-treatment combinations. Single-end sequencing was performed on the nine samples by the Illumina HiSeq™ 2000 platform, and raw sequence data generated is available for download at the NCBI Sequence Read Archive under number SRR4169965.
Functional annotation of the D. nobile transcriptome
After filtering of raw reads, de novo assembly of the transcriptome was carried out with Trinity14, a short reads assembly program. The generated unigenes were used for functional annotation against databases, including non-redundant (nr), SwissPort, COG, KEGG, and GO with a cut-off E-value of 0.00001.
Differential gene expression (DGE) analysis with D. nobile transcriptome
Total RNA extracted from four organs (i.e., leaf, pseudobulb, root, and axillary buds) at different time from one treatment were mixed at an equal mass ratio, and then acquired 3 RNA samples from cold-treated, TDZ-treated and control plants, respectively. The samples were used for RNA sequencing analysis via Illumina HiSeq™ 2000. After removal of low quality tags, empty tagsand tags with only one copy number, the raw DGE data (CK, LT, and TDZ),which were deposited in the GeneBank Short Read Archive (accession number SRR4266569), were mapped to the do novo assembled transcriptome of D. nobile. The gene expression level is calculated by using RPKM39 method, and the genes that were differentially expressed between the two samples were identified according to methods described by Audic and Claverie23.The false discovery rate (FDR) was used to determine the threshold P-value in multiple tests. We used FDR < 0.001 and an absolute value of the log2 ratio > 1 as the threshold to determine significant differences in gene expression. Differentially expressed genes were used for GO and KEGG enrichment analyses with a corrected P-value ≤ 0.05 as a threshold. Cluster analysis of gene expression patterns was performed with Cluster40 and Java Treeview41 software.
Gene expression variety analyzed by quantitative real-time PCR
A 500 ng sample of total RNA from axillary budswas used for reverse transcription using Quanta qScript cDNA SuperMix, and the cDNA was diluted 20 times before it was used for analysis by quantitative real-time PCR (qPCR). Each reaction consisted of 2 μl 20-fold cDNA, 9 μl SYBR-Green supermix, 1 μl of 6 μM forward and reverse primers, and 7 μl of sterile water. qPCR was performed in an Eppendorf Mastercycler ep realplex with the following parameters: one cycle of 2 min at 95 °C, followed by 40 cycles of15 s at 95 °C, 45 s at 57 °C, and 35 s at 68 °C. The last step for each reaction was a melting curve generation to test amplicon specificity. All qPCR reactions were performed in three technical and three biological replicates. All samples were compared with the control gene DnActin. The primer sequences for genes that were verified through qPCR are presented in Supplementary Table S1.
Additional Information
How to cite this article: Wen, Z. et al. Comparative Transcriptomic Analysis of Vernalization- and Cytokinin-Induced Floral Transition in Dendrobium nobile. Sci. Rep. 7, 45748; doi: 10.1038/srep45748 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Sung, S. & Amasino, R. M. Vernalization in Arabidopsis thaliana is mediated by the PHD finger protein VIN3. Nature 427(6970), 159–164 (2004).
Gendall, A. R., Levy, Y. Y., Wilson, A. & Dean, C. The VERNALIZATION 2 gene mediates the epigenetic regulation of vernalization in Arabidopsis. Cell 107(4), 525–535 (2001).
Levy, Y. Y., Mesnage, S., Mylne, J. S., Gendall, A. R. & Dean, C. Multiple roles of Arabidopsis VRN1 in vernalization and flowering time control. Science 297(5579), 243–246 (2002).
Bernier, G. My favourite flowering image: the role of cytokinin as a flowering signal. Journal of Experimental Botany, doi: 10.1093/jxb/err114 (2014).
Tarkowská, D. et al. Cytokinins in shoot apices of Brassica napus plants during vernalization. Plant Science 187(4), 105–112 (2012).
D’Aloia, M. et al. Cytokinin promotes flowering of Arabidopsis via transcriptional activation of the FT paralogue TSF. Plant Journal 65(6), 972–979 (2011).
Wong, C. E., Singh, M. B. & Bhalla, P. L. The dynamics of soybean leaf and shoot apical meristem transcriptome undergoing floral initiation process. Plos One 8(6), e65319–e65319 (2013).
Peter, B. Systematics of dendrobiinae (orchidaceae), with special reference to australian taxa. Botanical Journal of the Linnean Society 166(2), 105–126 (2011).
Leonhardt, K. W. Potted, blooming dendrobium orchids. Horttechnology 10(3), 431–432 (2000).
Rotor, G. B. Daylength and temperature in relation to growth and flowering of orchids. Cornell Experiment Station Bulletin 885, 3–47 (1952).
Rotor, G. B. The photoperiodic and temperature responses of orchids in The orchids: a scientific survey (ed. Withner, C. L. ) 397–416 (Ronald Press, 1959).
GOH, C. J. Hormonal regulation of flowering in a sympodial orchid hybrid Dendrobium louisae. New Phytologist 82(2), 375–380 (2006).
Wen, Z., Liu, Y., Lin, S., Lin, Y. & Liu, W. Cloning and expression analysis of a vrn1-like gene from Dendrobium nobile . Journal of South China Normal University 45(4), 109–114 (2013).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nature Biotechnology 29(7), 644–652 (2011).
He, Y. Control of the transition to flowering by chromatin modifications. Molecular Plant 2(4), 554–564 (2009).
Shannon, S. & Meeks-Wagner, D. R. A mutation in the Arabidopsis TFL1 gene affects inflorescence meristem development. Plant Cell 3(9), 877–892 (1991).
Alexandre, C. M. & Hennig, L. FLC or not FLC: the other side of vernalization. Journal of Experimental Botany. 59(6), 1127–1135 (2008).
Kim, D. H., Doyle, M. R., Sung, S. & Amasino, R. M. Vernalization: winter and the timing of flowering in plants. Annual Review of Cell and Developmental Biology 25, 277–299 (2009).
Liang, S. et al. Transcriptional regulations on the low-temperature-induced floral transition in an orchidaceae species, Dendrobium nobile: an expressed sequence tags analysis. Comparative & Functional Genomics 2012(1), 757–801 (2012).
Putterill, J., Robson, F., Lee, K., Simon, R. & Coupland, G. The CONSTANS gene of Arabidopsis promotes flowering and encodes a protein showing similarities to zinc finger transcription factors. Cell 80(6), 847–57 (1995).
Mcgaw, B. A. Cytokinin Biosynthesis and Metabolism in Plant Hormones and their Role in Plant Growth and Development (ed. Davies, P. J. ) 24–42 (M.Nijhoff, 1987).
Zürcher, E. & Müller, B. Cytokinin synthesis, signaling, and function—advances and new insights. International Review of Cell & Molecular Biology 324, 1–38 (2016).
Audic, S. & Claverie, J. M. The significance of digital gene expression profiles. GenomeResearch 7(10), 986–95 (1997).
Boggio, R. & Chiocca, S. Gam1 and the SUMO pathway. Cell Cycle 4(4), 533–5 (2005).
Wahl, V. et al. Regulation of flowering by trehalose-6-phosphate signaling in Arabidopsis thaliana . Science 339(6120), 704–7 (2013).
Silva, J. A. T. D., Aceto, S., Liu, W., Yu, H. & Kanno, A. Genetic control of flower development, color and senescence of Dendrobium orchids. Scientia Horticulturae 175(1), 74–86 (2014).
Cai, J. et al. The genome sequence of the orchid phalaenopsis equestris . Nature Genetics 47(1), 65–72 (2014).
Yan, L. et al. The genome of Dendrobium officinale illuminates the biology of the important traditional Chinese orchid herb. Molecular Plant 8, 922–934 (2014).
Chang, Y. Y. et al. Characterization of Oncidium “gower ramsey” transcriptomes using 454 GS-FLX pyrosequencing and their application to the identification of genes associated with flowering time. Plant & Cell Physiology 52(9), 1532–45 (2011).
Zhang, J. et al. Transcriptome analysis of cymbidium sinense and its application to the identification of genes associated with floral development. BMC Genomics 14(1), 1–17 (2013).
Su, C. L. et al. Orchidstra: an integrated orchid functional genomics database. Plant & Cell Physiology 54(2), e11–e11(2013).
Yang, W. et al. Regulation of meristem morphogenesis by cell wall synthases in arabidopsis. Current Biology Cb 26(11), 1404 (2016).
Kushwah, S. & Laxmi, A. The interaction between glucose and cytokinin signal transduction pathway in Arabidopsis thaliana . Plant Cell & Environment 37(1), 235–253 (2014).
Krucoff, M. et al. Florigenic effect of gibberellin on flowering according to period of chilling treatment in lavandula×intermedia . Horticulture Journal 85(2), 169–176 (2015).
Li, M. Z. et al. DELLA proteins interact with FLC to repress flowering transition. Journal of Integrative Plant Biology 58(7), 642–655 (2016).
Yamaguchi, N. et al. Gibberellin acts positively then negatively to control onset of flower formation in Arabidopsis . Science 344(6184), 638–641 (2014).
Moon, J. et al. The SOC1 mads‐box gene integrates vernalization and gibberellin signals for flowering in Arabidopsis . Plant Journal 35(5), 613–23 (2003).
Han, Y., Zhang, C., Yang, H. & Jiao, Y. Cytokinin pathway mediates apetala1 function in the establishment of determinate floral meristems In Arabidopsis. PNAS 111(18), 6840–6845 (2014).
Mortazavi, A., Williams, B. A., Mccue, K., Schaeffer, L. & Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-seq. Nature Methods 5(7), 621–628 (2008).
Sturn, A., Quackenbush, J. & Trajanoski, Z. Genesis: cluster analysis of microarray data. Bioinformatics 18(1), 207–208 (2002).
Clamp, M., Cuff, J., Searle, S. M. & Barton, G. J. The jalview java alignment editor. Bioinformatics 20(3), 426–427 (2004).
Acknowledgements
This research was supported by National Natural ScienceFoundation of China (30471223), Guangdong Natural Science Foundation (04020574), Science and Technology Planning Project of Guangdong Province (2003C201023), Agricultural science and technology project of Guangzhou (GK0401004) and Science and Technology Planning Project of Dongguan (2008B020400006).
Author information
Authors and Affiliations
Contributions
Z.W., W.L. and Q.Z. designed the experiments. J.L., W.G., H.L., and C.H. operated the experiments. Z.W. and Y.L. carried out data analysis and interpretation. Z.W., W.L. and Q. Z. wrote the paper. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Wen, Z., Guo, W., Li, J. et al. Comparative Transcriptomic Analysis of Vernalization- and Cytokinin-Induced Floral Transition in Dendrobium nobile. Sci Rep 7, 45748 (2017). https://doi.org/10.1038/srep45748
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep45748
This article is cited by
-
Flowering and flowering genes: from model plants to orchids
Horticulture, Environment, and Biotechnology (2021)
-
Exploring Flowering Genes in Isabgol (Plantago ovata Forsk.) Through Transcriptome Analysis
Plant Molecular Biology Reporter (2021)
-
Comparative transcriptome analysis of nonchilled, chilled, and late-pink bud reveals flowering pathway genes involved in chilling-mediated flowering in blueberry
BMC Plant Biology (2018)
-
Genome-wide researches and applications on Dendrobium
Planta (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
