
Abstract
Gut microbiota plays an important role in host health and nutrient digestion of animals. Probiotics have become one of effective alternatives to antibiotics enhancing animal health and performance through modulating gut microbiota. Previously, our research demonstrated that dietary Enterococcus Faecalis UC-100 substituting antibiotics enhanced growth and health of weaned pigs. To investigate the alterations of microbiota in the distal gut of pigs fed E. faecalis UC-100 substituting antibiotics, this study assessed fecal microbiota in pigs from different dietary treatments: the basal diet group, the E. faecalis group, and the antibiotic group on d 0, 14, and 28 of feeding through 16 S rRNA sequencing. Twenty-one phyla and 137 genera were shared by all pigs, whereas 12 genera were uniquely identified in the E. faecalis group on d 14 and 28. Bacterial abundance and diversity in the E. faecalis group, bacterial diversity in the antibiotic group, especially abundances of Fibrobacteres phylum and 12 genera in the E. faecalis group and antibiotics group were lower than that in the basal diet group on d 28. These results showed that microbial shifts in the porcine gut in response to diets containing E. faecalis were similar to the response to which containing antibiotics.
Similar content being viewed by others


Introduction
The large and diverse gut microbiota plays an important role in nutrient digestion and health of the host1,2,3,4. Weaning process always accompanies changes in intestinal structure and gut micro-ecosystem causing digestion dysfunction, diarrhea and growth inhibition in weaned pigs. Antibiotics have been used in pig production especially for weaned pigs to treat infectious diseases as well as to promote growth for more than 50 years4. However, the excessive use of antibiotics has caused the emergence of resistance of pathogenic bacteria to antimicrobials and a direct threat to human and animal health5,6,7. Therefore, efforts to find alternatives to antibiotics are being implemented to preserve the efficacy of current antimicrobials.
Probiotics and their metabolites have been suggested as the most desirable alternatives to support animal health, and as an effective way to promote growth through modulating gut microbiota in livestock2,8,9,10. Seeking effective probiotic bacteria as an alternative to antibiotics and untangling how these probiotics might affect host intestinal microbiota and immunity to improve the health and performance are essential steps for the successful application of probiotics in pig production11.
Enterococcus faecalis (E. faecalis), a lactic acid bacterium and common inhabitant in the gut12,13, is one of the most common species of Enterococci. Enterococci species including Enterococcus faecium strains have been studied for possible use as a probiotic and have shown beneficial effects on the health in pigs14,15,16. Some E. faecalis strains have been found beneficial to mice and humans improving their immunity17,18 and inhibiting pathogenic infection by producing bacteriocin19,20,21,22. However, limited studies are available demonstrating the application of the E. faecalis strains as alternatives to antibiotics in pig production23,24.
Previously, our research25 showed that weaned pigs fed a diet supplemented with E. faecalis UC-100 (200 g/t, ≥ 1 × 1010 CFU/g) had an increased body weight gain (increased by 30.9% on d 28 of feeding) and a decreased feed to gain ratio (decreased by 9.2% on d 28 of feeding) and incidence of diarrhea (decreased by 58.3% on d 28 of feeding) compared with weaned pigs fed a basal diet, whereas no difference was observed compared with weaned pigs fed a diet supplemented with antibiotics (bacitracin zinc: 40 g/t, aureomycin: 75 g/t and colistin: 20 g/t). However, specific mechanisms of the action for E. faecalis UC-100 were not clear. Considering that gut microbiota plays an important role in nutrient digestion and health of the host animals1,2,3,4, and many probiotics can promote animal growth and health through modulating gut microbiota2,8,9,10, it is hypothesized that E. faecalis UC-100 exerts growth and health promoting effect by altering gut microbiota in pigs. The objective of this study was to investigate alterations of microbiota in the porcine distal gut in response to dietary treatment of E. faecalis UC-100 as alternatives to antibiotics.
Results
DNA sequence data and quality control
A total of 2,355,992 paired-end 250-bp reads were acquired. The total read length was 1.97 gigabases (GB), and the average read length per sample was 0.05 GB. On d 0, 14, and 28 of feeding, there were 201,303, 212,206, and 222,669 raw reads in pigs from the basal diet group; 222,402, 217,627, and 216,349 raw reads in pigs from the E. faecalis group; 196,387, 207,015, and 215,410 raw reads in pigs from the antibiotic group, respectively (Table S1).
After quality control, 1,846,755 high quality sequences were obtained. On average, 51,298 sequences were obtained per sample. On d 0, 14, and 28 of feeding, there were 6,073, 6,378, and 6,908 operational taxonomic units (OTUs) in pigs from the basal diet group; 6,032, 6,459, and 6,235 OTUs in pigs from the E. faecalis group; 5,446, 5,856, and 6,274 OTUs in pigs from the antibiotic group, respectively, based on 97% species similarity (Table S1). A total of 9,966 OTUs were identified from all fecal samples (Table S1). Most OTUs were shared among groups at the same age, only 953 and 749 OTUs were uniquely identified in pigs from the E. faecalis group on d 14 and 28 of feeding, respectively (Fig. S1A). In addition, 753 and 729 OTUs were uniquely identified in pigs from the antibiotic group on d 14 and 28 of feeding, respectively (Fig. S1B).
Shifts in microbial abundance and diversity after E. faecalis treatment
Good’s coverage was at least 96% for each group. The range of the calculated value for Ace value was 5,887-7,482 over the 3 sampling times (d 0, 14, and 28 of feeding). On d 28 of feeding, the bacterial abundance in pigs from the E. faecalis group was lower (P < 0.01) than those from the basal diet group, whereas there was no difference between the E. faecalis group and the antibiotic group (Fig. 1A). And no difference in bacterial abundance was detected between the antibiotic group and the basal diet group on either d 14 or 28 of feeding (Fig. 1A). Compared with d 0 of feeding, bacterial abundance in pigs from the basal diet group and the antibiotic group increased (P < 0.01) on d 28, whereas bacterial abundance in the E. faecalis group increased (P < 0.05) on d 14 (Fig. 1B). No difference in bacterial diversity was detected among 3 groups on either d 14 or 28 of feeding (Fig. 1C). Compared with d 0 of feeding, bacterial diversity increased (P < 0.05) on d 28 from the basal diet group, whereas no change in the E. faecalis group and the antibiotic group (Fig. 1D). Weighted UniFrac distances were used to estimate β-diversity and to compare the three diet groups on d 14 (Fig. 2A,B and C) and d 28 of feeding (Fig. 2D,E and F). The PCoA plot of the weighted UniFrac distances showed that the three diet groups did not form distinct clusters on either d 14 or 28 of feeding, although the antibiotic group microbiota tended (P = 0.063) to separate from the Enterococcus faecalis group microbiota along principal coordinate 3 on d 14 (Fig. 2B and C).

The number of observed OTUs sharing ≥ 97% nucleotide sequence identity. Bacterial abundance was reflected with Ace index and bacterial diversity was reflected with OTUs number. (A) Ace index was compared among the basal diet group, the Enterococcus faecalis group and the antibiotic group at 3 different phases (d 0, 14, and 28 of feeding), respectively. (B) Ace index was compared among 3 different phases in the basal diet group, the Enterococcus faecalis group and the antibiotic group, respectively. (C) OTUs number was compared among the basal diet group, the Enterococcus faecalis group and the antibiotic group at 3 different phases, respectively. (D) OTUs number was compared among 3 different phases in the basal diet group, the Enterococcus faecalis group and the antibiotic group, respectively. (*p < 0.05, **p < 0.01).

PCoA of the weighted UniFrac distances for three groups on d 14 (A,B and C) and d 28(D,E and F) of feeding. The percent variation explained by each principal coordinate (A,B,C,D,E and F) is indicated on the axes.
Shifts in community membership after E. faecalis treatment
A total of 21 phyla were shared by pigs from all groups (Fig. S2 A), as follows: Actinobacteria, Bacteroidetes, Caldiserica, Chlamydiae, Chloroflexi, Crenarchaeota, Cyanobacteria, Deferribacteres, Euryarchaeota, Fibrobacteres, Firmicutes, Fusobacteria, Lentisphaerae, Planctomycetes, Proteobacteria, Spirochaetes, Synergistetes, Tenericutes, Thermi, TM7, and Verrucomicrobia. Of them, Firmicutes was the most dominant among the 21 phyla (P < 0.01) in the samples, and comprised more than 85% of the total sequences. Tenericutes and Bacteroidetes were the 2nd and 3rd dominant phyla which comprised only 1% of the total sequences. The bacterial abundance of Fibrobacteres in pigs from the E. faecalis group was lower (P < 0.01) than that in the basal diet group on d 28 of feeding, while no difference in bacterial abundance of Fibrobacteres was detected between the E. faecalis group and the antibiotic group (Fig. 3A,B). Meanwhile, the bacterial abundance of Fibrobacteres in pigs from the antibiotic group tended (P = 0.063) to be lower than that in the basal diet group on d 28 of feeding (Fig. 3A).

(A) The abundances of Fibrobacteres was compared among 3 different treatments on d 0, 14, and 28 of feeding, respectively. (B) The abundances of Fibrobacteres was compared among 3 different phases of experiment in the basal diet group, the Enterococcus faecalis group and the antibiotic group, respectively.
At the genus level, a total of 137 genera were identified from all samples (Fig. S2 B). The 11 most abundant genera, containing more than 85% of the total sequences, were Lactobacillus, Bulleidia, Clostridium, Streptococcus, Chlamydia, Coprococcus, Oscillospira, Eubacterium, Treponema, Ruminococcus, and Blautia. Of them, Chlamydia was a member of the phylum Chlamydiae, Treponema belongs to the phylum Spirochaetes and the other 9 genera belong to the phylum Firmicutes. Among the 11 most abundant genera, Lactobacillus, Bulleidia, and Clostridium were the most predominant genera, accounting for 23, 13, and 11% of total sequences, respectively. Most genera were shared among 3 groups at the same age, only 9 genera (Anaerovorax, Brevibacterium, Deinococcus, Facklamia, Ignatzschineria, Mycoplasma, Pedobacter, Sphingobium and Vibrio) and 3 genera (Burkholderia, Paraprevotella and Stenotrophomonas) were uniquely identified in pigs from the E. faecalis group on d 14 and 28 of feeding, respectively (Table 1). Only 10 (Acetobacter, Aequorivita, Anaerococcus, B-42, Holdemania, HTCC, Mycobacterium, Sporanaerobacter, Tsukamurella and Veillonella) and 1 (Ramlibacter) genera were uniquely identified in pigs from the antibiotic group on d 14 and 28 of feeding, respectively (Table 2).
The abundance of 12 genera (Acholeplasma, Arcobacter, Caldicoprobacter, Desulfotomaculum, Ignatzschineria, KSA1, Leptolyngbya, Natronincola_Anaerovirgula, Pseudomonas, Pseudoramibacter_Eubacterium, Tepidimicrobium, and Tissierella_Soehngenia) in pigs from both the E. faecalis group and the antibiotic group were lower (P < 0.05) than that in the basal diet group on d 28 of feeding, while no difference in bacterial abundance of these genera were detected between the E. faecalis group and the antibiotic group (Tables 3,4). The abundance of 5 genera (Anaerococcus, Fibrobacter, Megasphaera, Selenomonas and Sharpea) changed (P < 0.05) in pigs from the E. faecalis group than that in the basal diet group, and the abundance of 4 genera (Bacillus, Sphaerochaeta, Vibrio and Zhouia) changed (P < 0.05) in pigs from the antibiotic group than that in the basal diet group on d 28 (Table 5). However, no difference in bacterial abundance of these 9 genera was detected between the E. faecalis group and the antibiotic group (Table 5).
Discussion
Dietary supplementation of E. faecalis strains, as a probiotic, has become one of effective alternatives to the use of antibiotics to increase health and growth performance of pigs24,25 as it has been shown that probiotics can affect gut microbiota which plays an important role in health and nutrient digestion in pigs1,2,3,4. Although many studies have examined the impact of antibiotics on the gut microbiota in pigs1,4,26,27,28,29, there is very little information on how consumed E. faecalis affect the entire porcine gut microbiota24,30. Because many E. faecalis strains can inhibit pathogen by producing bacteriocin19,20,21,22, we try to study if E. faecalis UC-100 administration could induce alteration of the gut microflora to directly or indirectly impact porcine health and performance. Meanwhile, it is important to explore if E. faecalis UC-100 administration could cause alteration of the composition or activity of the host normal microbiota to exclude the possibility of the occurrence of undesirable microbiota changes before we applicate the E. faecalis UC-100 in pig production.
Since dietary supplementation of 200 g/t E. faecalis UC-100 showed the benefits similar to antibiotics supplementation from the previous manuscript25, samples from the E. faecalis UC-100 group(200 g/t), the basal diet group and the positive control diet group collected on d 0, 14, and 28 of feeding were used to determine alteration of the distal gut microbiota population in response to the treatment with E. faecalis substitute for antibiotics. We obtained 1,846,755 high-quality sequences from all samples, and the read counts were greater than those in previous studies in pigs1,4,28. Moreover, according to Good’s coverage index (96%) of each sample, the modified sequences were comprehensively enough to cover most bacterial diversity.
Venn diagrams were generated to make qualitative comparisons among E. faecalis group, antibiotic group and basal diet group at the same age. Most of the OTUs were shared between groups at the same age (Fig. S1A), which indicates that unique OTUs to each group were more likely to be found as less abundant OTUs, and this result is consistent with previous study1. In the basal diet group, bacterial abundance and diversity were increased with age, and these results are in accordance with a previous study31. Whereas, the increase of bacterial diversity and abundance were inhibited in pigs from the E. faecalis group compared with the basal diet group on d 28 of feeding. Mechanisms whereby E. faecalis UC-100 decrease bacterial diversity and abundance may be that E. faecalis strains can produce bacteriocin to inhibit pathogen and modulate other gut microbiota19,20,21,22. Although the increase of bacterial diversity was inhibited in pigs from the antibiotic group compared with the basal diet group on d 28 of feeding, no difference in bacterial abundance and diversity was detected between the antibiotic group and the basal diet group, and the addition of antibiotic to the swine diet did not shift the overall microbial community structure (β-diversity indices) on either d 14 or 28 of feeding. These results were similar to a previous study which showed that overall microbial community structure, microbial abundance and diversity in weaned pigs were not affected by chlortetracycline treatment28. Poole et al.32 also found no significant effect on a-diversity when pigs were fed with chlortetracycline for 28 days. In contrast, Looft et al.33 observed a significant decrease in total OTUs and the Shannon index indices in the early period (up to 4 days) of administration of carbadox in 6-weeks-old piglets, and Looft et al.4,27 also observed significant changes in microbial community structure (β-diversity indices) with antibiotic treatment. The discrepancies between the present study and the previous work4,27,33 might be resulted from the use of different type, dosage of antibiotics, the different environmental conditions or pig ages.
Firmicutes, Tenericutes and Bacteroidetes were the most dominant phyla in this study. These results were similar to previous studies1,4,31 where they showed that Firmicutes and Bacteroidetes were the most dominant phyla in pig fecal samples. Here we showed that E. faecalis UC-100 administration had no significant effect on proportions of dominant phyla. However, on d 28 of feeding, dietary E. faecalis UC-100 decreased the abundance of Fibrobacteres phyla. Fibrobacteres is a small and normal bacterial phylum which benefits the host by fermenting dietary fiber into short-chain fatty acids (SCFAs)34. Meanwhile, dietary antibiotics also tend to decrease the bacterial abundance of Fibrobacteres on d 28. These results are similar to results of a previous study where the authors showed that weaned pigs treated with tylosin had a lower proportion of Fibrobacteres sequences than those in the control group28. O’Toole PW et al.35 reported that consumption of probiotic may modulate the microbiota by competing for nutritional substrates, and by altering the dynamics of carbohydrate utilization by individual microbiota components. It can be speculated that E. faecalis UC-100 may modulate the Fibrobacteres by competing for nutritional substrates such as cellulose.
Lactobacillus, Bulleidia and Clostridium were the most predominant genera in the present study. These results are similar to a previous study where they showed that Lactobacillus and Clostridium were the most dominant genera in pig fecal samples31. Although most genera were shared among groups at the same age, 12 genera were uniquely identified in pigs from the E. faecalis group on d 14 or 28 of feeding. Of these unique genera, Anaerovorax functions to reduce the susceptibility to Campylobacter infection in humans36, Deinococcus is safely used as a feed supplement for hens37, and Paraprevotella may contribute to host health38. Conversely, it was found that Achromobacte and Gemella were specific to the basal diet group on d 14 and 28 of feeding, and several species of these 2 genera are opportunistic pathogens that affect humans39,40. In addition, 11 genera including Veillonella were uniquely identified in pigs from the antibiotic group on d 14 or 28 of feeding. Some Veillonella species have the function of utilization of macro- and micro-nutrients and may contribute to the regulation of host metabolism and body weight in human gut41. The existence of the unique beneficial genera in the E. faecalis group or the antibiotic group and the unique opportunistic pathogens in basal diet group may be a potential factor related to decreased incidence of diarrhea and increased body weight gain in the E. faecalis group and the antibiotic group.
Moreover, it was found that the bacterial abundance of 12 genera were increased as pigs aged in the basal diet group, but decreased in both the E. faecalis and antibiotic group on d 28. Of these 12 genera, many species of Pseudomonas42, Acholeplasma43, Arcobacter44,45,46, and Eubacterium47 are opportunistic pathogens that affect humans and animals. Several species of Pseudomonas42 and Arcobacter46 infections can cause diarrhea. Both antibiotic1 and E. faecalis19,20,21,22,23 can reduce or inhibit the presence of opportunistic pathogens, and this may be the reason that the bacterial abundance of these genera decreased and then caused the decreased incidence of diarrhea and the increased body weight gain in both E. faecalis UC-100 and antibiotic group. And the microbial shifted in the porcine gut in response to diets fed E. faecalis were similar to the response to dietary supplementation of antibiotics, indicating that E. faecalis UC-100 could be a potential alternative to the use of antibiotics in pigs to promote health and growth of host.
In addition, the bacterial abundance of 4 genera including Fibrobacter and Megasphaera were decreased, and Selenomonas were increased only in the pigs from the E. faecalis group compared with the basal diet group on d 28. A decrease in Fibrobacter genus was consistent with the decrease in Fibrobacteres phyla in E. faecalis group on d 28 of feeding. Some species of Megasphaera may cause diarrhea48, and Selenomonas was related to obesity49. The decrease of Megasphaera and the increase of Selenomonas may cause the decreased incidence of diarrhea and the increased body weight gain in the E. faecalis group. Meanwhile, the bacterial abundance of Bacillus and Sphaerochaeta, were decreased and the bacterial abundance of Vibrio and Zhouia were increased only in pigs from the antibiotic group compared with the basal diet group. Some species of Bacillus may cause cutaneous, gastrointestinal, and inhalation anthrax50, its decreased abundance may cause the decreased incidence of diarrhea in the antibiotic group. Previous study51 showed that bacteria of Vibrio might lead to development of acute gastroenteritis characterized by diarrhea, headache, vomiting, nausea, and abdominal cramps, its increased abundance in the antibiotic group might be due to its resistance to antibiotic52.
It was interesting to note that administration of E. faecalis UC-100 did not increase the abundance of Enterococcus genus. The lack of an effect on Enterococcus genera is probably due to the insufficient contribution of the Enterococcus faecalis strain, as E. faecalis UC-100 after intake still accounted for a minor part of the Enterococcus community in our samples.
Conclusion
The abundance and diversity of the gut microbiota in pigs of the E. faecalis group and the bacterial diversity in the antibiotic group were inhibited on d 28 of feeding. Most genera were shared among groups at the same age, 12 and 11 genera were uniquely identified in pigs from the E. faecalis group and the antibiotic group on d 14 or 28 of feeding, respectively. Several species of these unique genera can be beneficial to host health. The abundance of Fibrobacteres phylum and 12 genera including Fibrobacter and some opportunistic pathogens in pigs from both the E. faecalis group and the antibiotic group were lower than that in the basal diet group on d 28 of feeding. These results showed that microbial shifts in the porcine gut in response to diets fed E. faecalis were similar to the response to dietary supplementation of antibiotics, indicating that E. faecalis can be a potential alternative to the use of antibiotics in pigs.
Materials and Methods
Probiotics
E. faecalis UC-100 (CGMCC No.1.0130) is certified as an animal feed additive by the Ministry of Agriculture in China. The commercial product (the viable count of 1 × 1010 CFU/g) was obtained from Sikefu Biotechnology CO., LTD (Beijing, China).
Animals and sample collection
This experiment was approved by Animal Care and Use Committee of Nanjing Agricultural University. All procedures and the use of animals were carried out in accordance with the Guide for the Care and Use of Laboratory Animals prepared by the Institutional Animal Care and Use Committee of Nanjing Agricultural University, Nanjing, China.
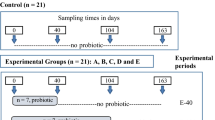
The experimental design and animal feeding procedure have been described previously25. Briefly, 150 newly weaned pigs (Duroc × Landrace × Yorkshire, 25 days of age at 8.4 ± 0.2 kg body weight, weaned at day 25) were allotted to 5 dietary treatments based on a randomized complete block design with gender and initial body weight as blocks. Each dietary treatment had 4 pens (replicates), and each pen had 7 or 8 pigs. Dietary treatments represent a basal diet, 3 test diets containing E. faecalis UC-100 at various levels (100, 200, and 400 g/t, respectively), or a positive control diet containing multiple antibiotics (bacitracin zinc 40 g/t, aureomycin 75 g/t, and colistin 20 g/t). Dietary treatments were given to pigs for 28 days. All pens were decontaminated and disinfected for 7 days before the pigs moved in to ensure minimal bacterial contamination. The building was temperature-controlled (26.3 ± 2 °C) during the study. Feed and water were available ad libitum for all pigs. Diet composition and nutrient contents are provided in the supplementary material (Table S2). The experiment was divided into 2 phases: phase I (from d 0 to d 14 of feeding) and phase II (from d 14 to d 28 of feeding). Fecal samples were collected from 1 randomly selected pig in all pens by rectal massage on d 0, 14 and 28 of feeding and then stored at −80 °C before DNA extraction. Each group had the same ratio between barrows and gilts. Because 200 g/t E. faecalis UC-100 showed the benefits similar to antibiotic supplementation from the previous manuscript25, only 36 fecal samples from the E. faecalis UC-100 group(200 g/t), the basal diet group and the positive control diet group collected on d 0, 14, and 28 of feeding were used in the current study based on the objective of investing microflora changes in the porcine distal gut in response to the treatment with E. faecalis UC-100 as alternatives to antibiotics. Gut microbiota population in fecal samples were assessed through 16 S rRNA gene sequencing.
DNA extraction, PCR amplification of 16 S rRNA gene, amplicon sequence and sequence data processing
Microbial genomic DNA was extracted from 220 mg of each fecal sample using a TIANamp Stool DNA Kit (Spin Column, Cat. no. DP328) according to the manufacturer’s recommendation. Successful DNA isolation was confirmed by agarose gel electrophoresis31.
The V4 hypervariable regions of 16 S rRNA gene were amplified by PCR using the barcoded fusion primers referred to previous study53. The primer sequences were 520 F 5-AYTGGGYDTAAAGNG-3 and 802 R 5-TACNVGGGTATCTAATCC-3. The PCR condition was as follows: initial denaturation at 94 °C for 4 min; 94 °C denaturation for 30 s, 50 °C annealing for 45 s, and 72 °C extension for 30 s, repeated for 25 cycles; final extension at 72 °C for 5 min. The PCR amplicon products were separated on 0.8% agarose gels and extracted from the gels. Only PCR products without primer dimers and contaminant bands were used for sequencing by synthesis. Barcoded V4 amplicons were sequenced using the paired-end method by Illumina MiSeq with a 7-cycle index read. Sequences with an average phred score lower than 30, ambiguous bases, homopolymer runs exceeding 6 bp, primer mismatches, or sequence lengths shorter than 100 bp were removed. Only sequences with an overlap longer than 10 bp and without any mismatch were assembled according to their overlap sequence. Reads that could not be assembled were discarded. Barcode and sequencing primers were trimmed from the assembled sequence31.
Taxonomy classification and sequence analysis
Taxon-dependent analysis was conducted using the Greengene database54. Greengenes is a quality controlled, comprehensive 16 S reference database and taxonomy based off a de novo phylogeny that provides standard operational taxonomic unit sets. OTUs were counted for each sample to express the richness of bacterial species with an identity cutoff of 97%. Low abundance OTUs (fewer than 5 reads) were filtered out of our analysis55. The OTU abundance of each sample was generated at genus level. The mean length of all effective bacterial sequences without primers was 227 bp. The abundance count at the genus level was log2 transformed and then normalized as follows: from each log-transformed measure, the arithmetic mean of all transformed values was subtracted, and the difference was divided by the standard deviation of all log-transformed values for a given sample. After this procedure, the abundance profiles for all samples exhibited a mean of 0 and a standard deviation of 1.
A Venn diagram was generated to compare OTUs between groups, and the bacterial community indices applied here included Ace and Good’s coverage. The bacterial abundance is shown by Ace. Good’s coverage estimates what percent of the total species is represented in a sample. The bacterial diversity is shown by the number of OTUs. β-diversity was calculated using weighted UniFrac distance and displayed using principal coordinate analysis (PCoA)28.
Data analysis
Only high-quality sequences obtained after quality control analysis were used in present analysis which were uploaded to QIIME for further study54. All effective bacterial sequences were compared to the Greengene databases using the best hit classification option to classify the abundance count of each taxon. The sequence length was archived by QIIME. The abundance and diversity indices were generated using Mothur with an OTU identity cutoff of 97% after implementing a pseudo-single linkage algorithm1. For all parameters, data were compared using a one-way analysis of variance (ANOVA) at the end of each bioassay. A mean comparison was performed using Fisher’s least significant difference test (LSD) and the Duncan multiple range test with a significance level of P < 0.05.
Additional Information
How to cite this article: Li, P. et al. Microbial shifts in the porcine distal gut in response to diets supplemented with Enterococcus Faecalis as alternatives to antibiotics. Sci. Rep. 7, 41395; doi: 10.1038/srep41395 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Kim, H. B. et al. Microbial shifts in the swine distal gut in response to the treatment with antimicrobial growth promoter, tylosin. Proc. Natl. Acad. Sci. USA. 109, 15485–15490 (2012).
Thacker, P. A. Alternatives to antibiotics as growth promoters for use in swine production: a review. J Anim Sci Biotechnol. 4, 35, 10.1186/2049-1891-4-35 (2013).
Hammesfahr, U. et al. Impact of the antibiotic sulfadiazine and pig manure on the microbial community structure in agricultural soils. Soil Biology and Biochemistry. 40, 1583–91 (2008).
Looft, T. et al. In-feed antibiotic effects on the swine intestinal microbiome. P Natl Acad Sci USA. 109, 1691–6 (2012).
Schwarz, S. & Chaslus-Dancla, E. Use of antimicrobials in veterinary medicine and mechanisms of resistance. Vet Res. 32, 201–225 (2001).
Phillips, I. et al. Does the use of antibiotics in food animals pose a risk to human health? A critical review of published data. J Antimicrob Chemother. 53, 28–52 (2004).
Gong, J., Yin, F., Hou, Y. & Yin, Y. Review: Chinese herbs as alternatives to antibiotics in feed for swine and poultry production: Potential and challenges in application. CAN J ANIM SCI. 94(2), 223–241 (2014).
Bednorz, C. et al. Feeding the Probiotic Enterococcus faecium Strain NCIMB 10415 to Piglets Specifically Reduces the Number of Escherichia coli Pathotypes That Adhere to the Gut Mucosa. Appl Environ Microbiol. 79, 7896–7904 (2013).
Roselli, M. et al. Alternatives to in-feed antibiotics in pigs: Evaluation of probiotics, zinc or organic acids as protective agents for the intestinal mucosa. A comparison of in vitro and in vivo results. Animal Research. 54, 203 (2005).
Meng, Q. et al. Influence of probiotics in different energy and nutrient density diets on growth performance, nutrient digestibility, meat quality, and blood characteristics in growing-finishing pigs. J Anim Sci. 88, 3320–6 (2010).
Butel, M. J. Probiotics, gut microbiota and health. Médecine et Maladies Infectieuses. 44, 1–8 (2014).
Gaggìa, F. et al. Probiotics and prebiotics in animal feeding for safe food production. Int J Food Microbiol. 141, 15–28 (2010).
Toit, M. D. et al. Preliminary characterization of bacteriocins produced by Enterococcus faecium and Enterococcus faecalis isolated from pig faeces. J Appl Microbiol. 88, 482–94 (2000).
Szabó, I. et al. Influence of a probiotic strain of Enterococcus faecium on Salmonella enterica serovar Typhimurium DT104 infection in a porcine animal infection model. Appl Environ Microb. 75, 2621–8 (2009).
Büsing, K. & Zeyner, A. Effects of oral Enterococcus faecium strain DSM 10663 NCIMB 10415 on diarrhoea patterns and performance of sucking piglets. Benef Microbes. 6, 41–44 (2015).
Bednorz, C. et al. Feeding the probiotic Enterococcus faecium strain NCIMB 10415 to piglets specifically reduces the number of Escherichia coli pathotypes that adhere to the gut mucosa. Appl Environ Microbiol. 79, 7896–7904 (2013).
Hoffmann, M. et al. Impact of a probiotic Enterococcus faecalis in a gnotobiotic mouse model of experimental colitis. Mol Nutr Food Res 55, 703–713 (2011).
Sparo, M. et al. Immunomodulatory properties of cell wall extract from Enterococcus faecalis CECT7121. Braz J Infect Dis. 18, 551–555 (2014).
Hugas, M. et al. Functionality of enterococci in meat products. Int J Food Microbiol. 88, 223–233 (2003).
Nilsen, T. et al. a cell wall-degrading bacteriocin from Enterococcus faecalis LMG 2333. Appl Environ Microbiol. 69, 2975–84 (2003).
Huang, E. et al. Characterization and application of enterocin RM6, a bacteriocin from Enterococcus faecalis. Biomed Res Int. 2013, 206917, 10.1155/2013/206917 (2013).
Liu, X. et al. Identification of an N-terminal formylated, two-peptide bacteriocin from Enterococcus faecalis 710C. J Agric Food Chem. 59, 5602–8 (2011).
Tsukahara, T. et al. Evaluation of the heat-killed and dried cell preparation of Enterococcus faecalis against villous atrophy in early-weaned mice and pigs. Anim Sci J. 82, 302–306 (2011).
Hu, Y. et al. Dietary Enterococcus faecalis LAB31 Improves Growth Performance, Reduces Diarrhea, and Increases Fecal Lactobacillus Number of Weaned Piglets. PLoS One. 10, e0116635, 10.1371/journal.pone.0116635 (2015).
Wei, Q. T. et al. Effect of dietary Enterococcus faecalis replacing of antibiotic on growth performance, diarrhea rate, humoral immunity and intestinal microflora of nursery pigs. J of Nanjing Agricultural University. 37, 143–148 (2014).
Allen H. K. et al. Antibiotics in feed induce prophages in swine fecal microbiomes. MBio. 2, e00260–11, 10.1128/mBio.00260-11 (2011).
Looft, T. et al. Bacteria, phages and pigs: the effects of in-feed antibiotics on the microbiome at different gut locations. ISME J. 8, 1566–1576 (2014).
Holman, D. B. & Chénier, M. R. Temporal changes and the effect of subtherapeutic concentrations of antibiotics in the gut microbiota of swine. FEMS Microbiol Ecol. 90, 599–608 (2014).
Kim, J. et al. Effects of the Antibiotics Growth Promoter Tylosin on Swine Gut Microbiota. J Microbiol Biotechnol. 26, 876–82 (2016).
Kong, X. F. et al. Dietary supplementation with chitooligosaccharides alters gut microbiota and modifies intestinal luminal metabolites in weaned Huanjiang mini-piglets. LIVEST SCI. 160(1), 97–101 (2014).
Niu, Q. et al. Dynamic distribution of the gut microbiota and the relationship with apparent crude fiber digestibility and growth stages in pigs. Sci Rep. 5, 9938, 10.1038/srep09938 (2015).
Poole T. et al. The effect of chlortetracycline on faecal microbial populations in growing swine. J Glob Antimicrob Resist. 1, 171–174 (2013).
Looft, T. et al. Carbadox has both temporary and lasting effects on the swine gut microbiota. Front Microbiol. 5, 276 (2014b).
Abdul Rahman N. et al. A Phylogenomic Analysis of the Bacterial Phylum Fibrobacteres. Front Microbiol. 6, 1469, 10.3389/fmicb.2015.01469 (2016).
O’Toole, P. W. & Cooney, J. C. Probiotic bacteria influence the composition and function of the intestinal microbiota. Interdiscip Perspect Infect Dis. 2008, 175285, 10.1155/2008/175285 (2008).
Dicksved, J. et al. Susceptibility to Campylobacter infection is associated with the species composition of the human fecal microbiota. MBio. 5, e01212–14, 10.1128/mBio.01212-14 (2014).
Wu, S. Y. et al. Characterization and safety evaluation of a Deinococcus member as feed additive for hens. Regul Toxicol Pharmacol. 76, 121–127 (2016).
Liu, J. et al. Acute cholecystitis associated with infection of Enterobacteriaceae from gut microbiota. Clin Microbiol Infect. 21, 851.e1–9, 10.1016/j.cmi.2015.05.017 (2015).
Cools, P. et al. Epidemic Achromobacter xylosoxidans strain among Belgian cystic fibrosis patients and review of literature. BMC Microbiol. 16, 122, 10.1186/s12866-016-0736-1 (2016).
Brouqui, P. et al. Endocarditis due to rare and fastidious bacteria. Clinical Microbiology Reviews. 14, 177–207 (2001).
Palleja A. et al. Roux-en-Y gastric bypass surgery of morbidly obese patients induces swift and persistent changes of the individual gut microbiota. Genome Med. 8(1), 67 (2016).
Mantareva, V. et al. Photodynamic inactivation of pathogenic species Pseudomonas aeruginosa and Candida albicans with lutetium (III) acetate phthalocyanines and specific light irradiation. Lasers Med Sci. 10.1007/s10103-016-2022-8 (2016).
Windsor, H. M. et al. The growth and long term survival of Acholeplasma laidlawii in media products used in biopharmaceutical manufacturing. Biologicals. 38, 204–210 (2010).
Vandenberg, O. et al. Arcobacter species in humans. Emerging Infectious Diseases. 10, 1863–1867 (2004).
Fera, M. T. et al. Detection of Arcobacter spp. in the coastal environment of the Mediterranean see. Appl and Environ Microbiol. 70, 1271–1276 (2004).
Ho, H. T. et al. Arcobacter, what is known and unknown about a potential foodborne zoonotic agent! Vet Microbiol. 115, 1–13 (2006).
Africa, Charlene . et al. Anaerobes and Bacterial Vaginosis in Pregnancy: Virulence Factors Contributing to Vaginal Colonisation. Int J of Environ Res Public Health. 11, 6979–7000 (2014).
Hashizume K. et al. Megasphaera elsdenii JCM1772 Normalizes Hyperlactate Production in the Large Intestine of Fructooligosaccharide-Fed Rats by Stimulating Butyrate Production. J Nutr, 133(10), 3187–3190 (2003).
Anjum J., Syed A. A. & Farah V. M. Obesity- Caused by a germ? IJSRP. 3(1), 2250–3153 (2013).
Spencer, R. C. Bacillus anthracis. J Clin Pathol. 56, 182–187 (2003).
Su, Y. C. & Liu, C. Vibrio parahaemolyticus: a concern of seafood safety. Food Microbiol. 24(6), 549–558 (2007).
Dubert J. et al. Persistence of Antibiotic Resistant Vibrio spp. in Shellfish Hatchery Environment. Microb Ecol. 72(4), 851–860 (2016).
Zhao, L. et al. Quantitative genetic background of the host influences gut microbiomes in chickens. Sci Rep. 3, 1163, 10.1038/srep01163 (2013).
McDonald, D. et al. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. The ISME journal. 6, 610–618 (2012).
Bokulich, N. A. et al. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat Methods. 10, 57–59 (2013).
Acknowledgements
This work was supported by the National Science and Technology Program for the Twelfth Five Year Plan for Rural Development (2015BAD03B02-4), the Sanxin Agricultural Engineering Project of Jiangsu Province (SXGC(2015)319, SXGC(2016)275, SXGC(2016)146). The sequencing service was provided by Personal Biotechnology Co., Ltd., Shanghai, China.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: R.H.H. P.H.L. Performed the experiments: P.H.L. Q.N. Q.T.W. Y.Q.Z. M.X.L. Analyzed the data: Q.N. P.H.L. Y.Q.Z. X.M. Contributed reagents/materials/analysis tools: P.H.L. Q.N. R.H.H. Q.T.W. Y.Q.Z. M.X.L. Contributed to the writing of the manuscript: P.H.L. Q.N. R.H.H. S.W.K. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Li, P., Niu, Q., Wei, Q. et al. Microbial shifts in the porcine distal gut in response to diets supplemented with Enterococcus Faecalis as alternatives to antibiotics. Sci Rep 7, 41395 (2017). https://doi.org/10.1038/srep41395
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep41395
This article is cited by
-
Growth performance, lipid metabolism, and systemic immunity of weaned piglets were altered by buckwheat protein through the modulation of gut microbiota
Molecular Genetics and Genomics (2024)
-
Pectin modulates intestinal immunity in a pig model via regulating the gut microbiota-derived tryptophan metabolite-AhR-IL22 pathway
Journal of Animal Science and Biotechnology (2023)
-
Administration of probiotic lactic acid bacteria to modulate fecal microbiome in feedlot cattle
Scientific Reports (2022)
-
An important link between the gut microbiota and the circadian rhythm: imply for treatments of circadian rhythm sleep disorder
Food Science and Biotechnology (2022)
-
Comparative analysis of the pulmonary microbiome in healthy and diseased pigs
Molecular Genetics and Genomics (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
