
Abstract
The sustainable production of fuels and chemicals using microbial cell factories is now well established. However, many microbial production processes are still limited in scale due to inhibition from compounds that are present in the feedstock or are produced during fermentation. Some of these inhibitors interfere with cellular membranes and change the physicochemical properties of the membranes. Another group of molecules is dependent on their permeation rate through the membrane for their inhibition. We have investigated the use of membrane engineering to counteract the negative effects of inhibitors on the microorganism with focus on modulating the abundance of complex sphingolipids in the cell membrane of Saccharomyces cerevisiae. Overexpression of ELO3, involved in fatty acid elongation, and AUR1, which catalyses the formation of complex sphingolipids, had no effect on the membrane lipid profile or on cellular physiology. Deletion of the genes ORM1 and ORM2, encoding negative regulators of sphingolipid biosynthesis, decreased cell viability and considerably reduced phosphatidylinositol and complex sphingolipids. Additionally, combining ELO3 and AUR1 overexpression with orm1/2Δ improved cell viability and increased fatty acyl chain length compared with only orm1/2Δ. These findings can be used to further study the sphingolipid metabolism, as well as giving guidance in membrane engineering.
Similar content being viewed by others
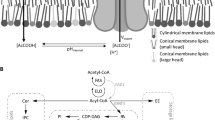

Introduction
Industrial processes based on microbial conversion have gained increased attention in the transformation to a more sustainable society. Microbial bioprocesses often have advantages over traditional chemical processes; for example, they make use of renewable feedstock, the catalysis conditions are more environmentally friendly, and it may be possible to produce new chemicals, difficult to synthesize using traditional chemistry1. A common limitation of microbial conversion is the inherent difficulty of the fermenting microorganism to maintain high productivity when conditions are perturbed2,3. For example, compounds present in the feedstock or that are produced during fermentation can negatively interfere with the physiology and key biochemical pathways of the microorganism4. One mode of inhibition through which these compounds can act is via interactions with biological membranes and change physicochemical properties of the membranes. A major problem in butanol production, for example, is the interaction of butanol with the plasma membrane of the fermenting organism, causing increased membrane fluidity, in turn leading to cofactor leakage and loss of membrane potential5. Ethanol has a similar mode of inhibition to that of butanol, although it is not as detrimental6. Inhibition by weak acids, on the other hand, is indirectly dependent on the plasma membrane lipid composition as they enter the cell mainly by passive diffusion over the plasma membrane, and inhibit vital functions once inside the cell7,8.
It may be possible to reduce or prevent inhibition due to molecules interacting with the cell membrane by engineering the lipid composition of the membrane. The permeability of the membrane could also be altered to reduce the uptake of inhibitory molecules to prevent damaging effects inside the cell. This concept, referred to as membrane engineering, is an attractive tool in cell factory design3, although only a limited number of membrane engineering projects have been described in the literature. In a study on Saccharomyces cerevisiae, it was shown that increasing the fraction of oleic acid (C18:1 fatty acid) in the plasma membrane improved the viability of the microorganism in the presence of inhibitory levels of ethanol, propanol and butanol9. The same strategy has also been used to increase the viability of yeast in the presence of acetic acid10. In another study, genetic modifications leading to enriched trans unsaturated fatty acids in the plasma membrane of Escherichia coli, improved its tolerance to octanoic acid as well as many alcohols, aromatic solvents and organic acids11.
We have previously demonstrated that the highly acetic acid tolerant yeast, Zygosaccharomyces bailii, has a unique ability to remodel the composition of its plasma membrane in response to acetic acid stress, greatly increasing the fraction of sphingolipids at the expense of glycerophospholipids12. Using in silico molecular dynamic simulations in combination with in vivo reduction of the sphingolipid fraction, we further demonstrated that the high sphingolipid content in the plasma membrane reduced the rate of acetic acid diffusion into the cell, resulting in increased acetic acid tolerance8.
Sphingolipid synthesis in S. cerevisiae requires three main building blocks: a long-chain base (LCB), a very-long-chain fatty acid (VLCFA, C24-C26), and an inositol-containing head group. A simplified illustration of the sphingolipid synthesis pathway is shown in Fig. 1. A LCB is formed through the condensation of serine and a fatty acyl-CoA (C16-C18) with the serine palmitoyltransferase (SPT) complex consisting of the two main proteins Lcb1 and Lcb2, and the smaller subunit Tsc313,14. A C18 LCB is formed by condensation of C16 fatty acid with serine followed by decarboxylation. The activity of the SPT complex is negatively regulated by the two paralogues Orm1 and Orm2, through physical interaction, as well as by Sac1, which negatively regulates the complex via an unknown mechanism15. A VLCFA is formed by a cyclic series of elongation reactions in the endoplasmic reticulum (ER) carried out by the fatty acid elongation complex, in which Elo1, Elo2 and Elo3 are elongases with affinity for different fatty acyl chains, but only Elo3 is able to synthesize the longest C26 fatty acids16. LCBs and VLCFAs are combined into ceramides by the action of ceramide synthase, consisting of one of the paralogues Lag1 or Lac1 together with Lip117. Phosphatidylinositol (PI) donates its inositol phosphate group to the ceramide moiety catalysed by the enzyme Aur1, forming inositol phosphoceramide (IPC). Mannosyl-inositol phosphoceramide (MIPC) is then formed by the addition of mannose to the inositol unit. In the last reaction step of sphingolipid synthesis, a second inositol phosphate from PI is added to MIPC, resulting in the formation of mannosyl-di-inositol phosphoceramide (MIP2C)18. IPC, MIPC and MIP2C are the sphingolipids located to the plasma membrane, and are collectively referred to as complex sphingolipids.

Genes overexpressed in this study are indicated in green and genes deleted are indicated in red. Abbreviations: VLCFA, very-long-chain fatty acid (C24-C26); LCB, long-chain base; PI, phosphatidylinositol; PI4P, phosphatidylinositol 4-phosphate; IPC, inositol phosphoceramide; MIPC, mannosyl-inositol phosphoceramide; MIP2C, mannosyl-diinositolphosphoceramide. The Illustration is drawn by the authors.
We propose membrane engineering as a tool to reduce the effects of inhibitors, and focus in the present study on the possibility of modulating the abundance of complex sphingolipids in the cell membrane of S. cerevisiae by overexpressing the genes ELO3 and AUR1 in combination with the deletion of the genes ORM1 and ORM2 (Fig. 1).
Materials and Methods
Strains and strain construction
S. cerevisiae strain CEN.PK 113_6B (MATa, SUC2, MAL2-8c, ura3-52, leu2, trp1)19 was used as the background strain. The constructed strains are listed and described in Table 1. All strains were constructed by integrating the changes into the yeast genome. The ELO3 and AUR1 gene sequences were amplified from genomic DNA of CEN.PK 113_6B using high-fidelity DNA polymerase (Thermo Fisher Scientific) and cloned into the integrative plasmids YIp211 and YIp128, respectively. The genes were constitutively expressed by the strong TEF2 promoter and terminated using CYC1. YIp211 harboured the URA3 marker, while YIp128 harboured the LEU2 marker. The plasmids were linearized using the restriction enzyme Stu1 or Cla1 cleaving the URA3 or LEU2 gene, to allow for homologous recombination at the ura3 or leu2 locus of the genome. For the control strains, empty plasmids harbouring only the marker gene were integrated into the marker locus. The plasmid constructs were verified by DNA sequencing before integration.
Deletion cassettes were constructed using high-fidelity DNA polymerase to amplify the TRP1 or KanMX marker from YIp204 or pUG6 plasmids. Primers with 20 bases binding to the marker sequence and 40 bases non-bonding tails, corresponding to the 5′ or 3′ ends respectively, of either the ORM1 or ORM2 gene were used. The cassettes were integrated at the locus of the gene to be deleted, looping out the gene and replacing it with the selected marker. For the control strain, only the TRP1 gene was amplified and integrated at the trp1 locus.
Yeast transformation was performed using the LiOAc method20, and transformants were selected on yeast nitrogen base (YNB) plates with complete supplement mixture (CSM) minus the appropriate dropout (6.9 g L−1 YNB with ammonium sulphate, 0.74 g L−1CSM-ura/leu/trp (MP Biomedicals), 20 g L−1 glucose, plates 20 g L−1 agar). Strains with the KanMX marker were selected on yeast extract peptone dextrose (YPD) plates supplemented with 0.2 g L−1 Geneticin (20 g L−1 peptone, 10 g L−1 yeast extract, 20 g L−1 glucose, 0.2 g L−1 Geneticin, 20 g L−1 agar). Correct integration into the yeast genome was verified by PCR on extracted genomic DNA. Four to six clones from each transformation were screened for growth in liquid medium, and the clone that displayed growth similar to that of the majority of the clones was selected for subsequent experiments.
Aerobic batch cultivation
Inoculum was prepared in Erlenmeyer flasks containing minimal medium: 20 g L−1 glucose, 5 g L−1 (NH4)2SO4, 0.5 g L−1 MgSO4 × 7H2O, 3 g L−1 KH2PO4, 1 mL L−1 vitamin solution (including 25 mg L−1 myo-inositol, final concentration) and 1 mL L−1 trace element solution. Vitamin solution and trace element solution were prepared as described previously21. The pH was maintained by the buffering capacity of 100 mM potassium hydrogen phthalate adjusted to pH 5 using KOH. The cultures were grown under continuous shaking at 180 rpm at 30 °C and occupied a maximum of 10% of the flask volume. Exponentially growing cells from overnight culture were harvested by centrifugation at 3000 × g for 3 min at room temperature, resuspended in fresh medium and added to the cultures at an initial optical density at 600 nm (OD600) of 0.2. Initial strain characterization of ELO3-OE, AUR1-OE and ELO3-AUR1-OE was performed in triplicate in 500 mL baffled Erlenmeyer flasks using a medium with the same composition and at the same culturing conditions as the inoculum.
Further characterization of the ELO3-AUR1-OE, orm1/2Δ and ELO3-AUR1-OE-orm1/2Δ mutants was carried out in triplicate in bioreactors (DASGIP AG, Jülich, Germany) with a 700 mL working volume, in minimal medium, as described above, supplemented with amino acids, 0.74 g L−1 CSM (MP Biomedicals) and 1.05 g L−1 L-serine. Serine was added due to a lack of serine in the CSM amino acid mixture. The serine concentration added corresponds to the approximate serine concentration in YPD medium22. Antifoam (0.1 μL mL−1 Antifoam 204, Sigma Aldrich) was also added to the growth medium to prevent foaming. The temperature was kept at 30 °C, and the pH was maintained at 5 by the addition of KOH. A constant airflow of 1 VVM was used to sparge the bioreactors, and the dissolved oxygen tension was maintained at a minimum of 40% by adjusting the stirrer speed throughout fermentation, using 400 rpm as the initial stirrer speed. The concentrations of oxygen and carbon dioxide in the inlet gas and in the off-gas were measured with on-line gas analysers (DASGIP AG, Jülich, Germany).
Quantification of glucose and extracellular metabolites
Samples withdrawn from bioreactor cultures were immediately filtered using 0.2 μm nylon filters. Samples were stored at −20 °C prior to analysis. Glucose and extracellular metabolites (acetate, ethanol, glycerol, pyruvate and succinate) were analysed using HPLC (Ultimate 3000, Dionex, Sunnyvale, CA, USA) together with an Aminex HPX87-H column (Bio-Rad Laboratories, Munich, Germany). 5 mM H2SO4 was used as the mobile phase, and the column temperature was maintained at 45 °C. Ethanol, glucose and glycerol were analysed using a refractive index detector (Shodex RI-101, Showa Denko, New York, NY, USA), while acetate, pyruvate and succinate were analysed using the UV detector on the Ultimate 3000 HPLC equipment, set at a wavelength of 210 nm. Six-point standard curves were used for quantification. Evaporation of ethanol was compensated for by assuming an evaporation rate of 1% ethanol per hour, of the ethanol present at each specific time point.
Quantification of dry cell weight
Dry cell weight was determined in duplicate by filtering approximately 10 g cell broth through dry pre-weighed 0.45 μm polyethersulfone (PES) membranes and then washing with deionized water (Sartorius Stedim, Aubagne, France). The exact weight of each sample was noted, and a density of 1 g mL−1 was assumed for the cell broth. The membranes were dried in a microwave oven at 120 W for 15 minutes and then placed in a silica gel desiccator for a minimum of 24 hours before weighing, to ensure that no moisture remained.
Cell viability
Cell viability was measured during mid-exponential growth by staining cells with methylene blue and counting the fraction of living cells using a microscope.
Gene expression analysis
Cell samples (5 mL) were collected from bioreactor cultivations during mid-exponential growth, immediately placed on ice and then harvested by centrifugation at 3000 × g for 3 min at 4 °C. The cell pellets were immediately frozen in liquid nitrogen and stored at −80 °C. The RNA was extracted at a later time using the RNeasy Mini kit (Qiagen) according to the manufacturer’s instructions. Adequate integrity of the extracted RNA was confirmed by gel electrophoresis. Reverse transcription of RNA to cDNA was performed using the RevertAid H Minus First Strand cDNA Synthesis kit (Thermo Fisher Scientific) with oligo (dT)18 primers, according to the manufacturer’s instructions. Control reactions to which no reverse transcriptase had been added (no RT) were also included.
Expression levels of ELO3 and AUR1 were analysed using quantitative real-time PCR (qPCR) using Brilliant II SYBR Green qPCR Master Mix (Agilent Technologies) and a Stratagene MX3005P qPCR instrument (Agilent Technologies). Primers amplifying the ELO3 and AUR1 gene segments were designed to avoid the formation of secondary structures of the amplicons and the primers using the Primer3 software23,24, see Table 2. qPCR reactions were prepared with 2 ng μL−1 cDNA template and 0.3 μM of each primer in 25 μL reaction volume. DNA was amplified using the following temperature profile: initial denaturation for 10 min at 95 °C, 40 cycles of amplification with 30 s denaturation at 95 °C and 1 min annealing/elongation at 60 °C. The specificity of the amplicons was then determined by dissociation of the DNA duplex by for 1 min at 95 °C, 1 min at 55 °C and 30 s at 95 °C. Three technical replicates of each of the three biological replicates were evaluated.
The MxPro software (Agilent Technologies) was used to analyse the data. Thresholds for quantification were set in the exponential region of the amplification curve, and the program determined the quantification cycle (Cq) based on the threshold for quantification. Expression levels of ELO3 and AUR1 were normalized to the expression of the TAF10 reference gene. Expression levels of the target genes were calculated as the relative expression between the mutant and the wild type using the 2−ΔΔCq method25. Control reactions without a template gave almost no amplification, while controls without reverse transcription (no RT) had a Cq value more than seven cycles higher than the Cq value of the target reaction, thereby indicating sufficiently low background levels to ensure no interference with the results. Four-point standard curves were used to determine the reaction efficiencies (101% for the ELO3 gene and 98% for the AUR1 gene), both of which are sufficient not to influence the calculated expression levels.
Lipid analysis
Cells corresponding to an OD600 of 25 were collected from bioreactor cultivations during mid-exponential growth and quenched by adding 100% (w/v) trichloroacetic acid (TCA) directly to the samples to give a final TCA concentration of 5% (w/v). Quenched samples were placed on ice for 10 minutes and then centrifuged at 3000 × g for 3 min at 4 °C to obtain a cell pellet. The cell pellet was then washed with 10 mL of 5% (w/v) TCA and then with 10 mL distilled water before freezing of the pellet at −80 °C until analysis.
Lipids were extracted by re-suspending the cell pellet in 3 mL extraction solvent containing ethanol, water, diethyl ether, pyridine and 4.2 N ammonium hydroxide (15:15:5:1:0.18, v/v). Internal standards were added (3.75 nmol PC31:1, 3.75 nmol PE31:1, 3.0 nmol PI31:1, 2.0 nmol PS31:1, 0.6 nmol C17Cer and 1 nmol C8GC) and glycerophospholipids and sphingolipids were thereafter extracted as described previously26.
Glycerophospholipids and sphingolipids were analysed using electrospray ionization multiple-reaction-monitoring mass spectrometry (ESI-MRM-MS) using a triple-stage quadrupole mass spectrometer (TSQ Vantage MS, Thermo Scientific, Whaltman, MA, USA) equipped with a robotic nanoflow ion source (Nanomate, Advion Biosciences, Ithaca, NY, USA). The different lipid species were quantified in relation to the appropriate internal standard and normalized to the total amount of phosphate in each sample. Three to six technical replicates were analysed for each of the three biological replicates. The results are expressed as apparent quantities in arbitrary units (a.u.). The arbitrary units of glycerophospholipids are comparable within the different classes, but are not comparable with the arbitrary units of sphingolipids. The absolute quantities of the three classes of sphingolipids could not be determined as no standards were available.
Statistical analysis
Statistical differences were tested between the constructed strains for each measured parameter. To account for multiple comparisons of strains, one-way ANOVA was selected as statistical test. To allow for pairwise comparisons, Turkey HSD was used in conjunction to the ANOVA analysis.
Results and Discussion
The possibility of modulating the abundance of complex sphingolipids in the cell membrane of S. cerevisiae towards the final goal of developing robust cell factories was investigated. To increase the flux through the sphingolipid biosynthesis pathway, VFLCA, LCB and IPC production were targeted by overexpression of the genes ELO3 and AUR1 in combination with the deletion of the genes ORM1 and ORM2 (Fig. 1). The engineered strains were evaluated in terms of cell physiology and cell lipid profile.
Overexpression of ELO3 and AUR1 causes no change in the lipid profile
In an attempt to increase the production of VLCFAs, the ELO3 gene encoding a protein in the fatty acid elongation complex required for the production of C26 fatty acids27, was overexpressed. AUR1 was also overexpressed in an attempt to increase the flux through the sphingolipid synthesis pathway, resulting in the accumulation of complex sphingolipids rather than the accumulation of the toxic intermediate ceramide. Initial characterization of the single and double mutants in aerobic shake flasks with buffered minimal medium revealed no effect on maximum specific growth rate, cell viability or metabolite profile (ethanol, glycerol, acetate, succinate, pyruvate) compared to the wild type (data not shown). Therefore, only the double mutant was selected for further characterization using pH- and oxygen-controlled bioreactors with minimal medium supplemented with amino acids. The ELO3-AUR1-OE mutant behaved similarly in this set-up, showing no significant effect on the fermentation profile compared to the wild type (Table 3). Investigation of the lipid profile, specifically the complex sphingolipid, ceramide and glycerophospholipid profile, revealed no significant difference between the ELO3-AUR1-OE mutant and the wild type (Fig. 2). To verify the overexpression of ELO3 and AUR1, the RNA levels of the specific genes were quantified using qPCR, which revealed a 1.9× average ((1.6–2.4)×, 95% CI) increase in RNA levels of ELO3, and a 5.2× average ((4.2–6.3)×, 95% CI) increase in RNA levels of AUR1.

(a) Complex sphingolipid classes. (b) Total ceramides. (c) Glycerophospholipid classes. (d) Degree of unsaturation per lipid in total glycerophospholipids. (e) Complex sphingolipid total chain length per lipid. (f) Ceramide chain length per lipid. (g) Total glycerophospholipid chain length per lipid. *Significant difference compared to wild type. #Significant difference compared with orm1/2Δ mutant. Differences obtained using one-way ANOVA with post-hoc Tukey HSD for multiple pairwise comparisons (p < 0.05). The results were calculated from three biological replicates and are given as the mean ± standard deviation.
The small impact of the ELO3-AUR1-OE modification was partly expected, since no measures had been taken in this strain to increase the LCB production. Therefore, even with an increased capacity to produce VLCFAs, ceramide synthesis and subsequent complex sphingolipid synthesis, promoted by the overexpression of AUR1, could not increase due to limited LCB availability.
The effect of ELO3 and AUR1 overexpression on the lipid profile has not been investigated previously, although lipid data showing the reduced expression of these genes are available in the literature. Strains with elo3Δ produce shorter acyl chains27,28,29, higher ceramide levels and lower levels of total complex sphingolipids than the wild type15,30. Similarly, abolished Aur1 activity resulted in increased ceramide accumulation and reduced total levels of complex sphingolipids31. However, reducing the flux through a metabolic pathway can be attained by completely suppressing the enzymatic activity of certain steps in the pathway and relatively easy create a different lipid profile. On the other hand, significantly increasing the flux can be challenging, as it is crucial to identify and target the steps in the pathway that exert the highest flux control, otherwise no effect will be seen. Limited effect of the overexpression of structural genes may also be due to decoupling of mRNA levels from the amount of active enzymes, post-translational modifications altering the enzyme activity, the low availability of other interacting enzymes, or feedback inhibition of the enzyme by the downstream products. Elo3 is part of a large enzyme complex32 and may be regulated by phosphorylation, as shown for its homologue Elo229. In addition, the activity of Aur1 has been shown to be dependent on the enzyme Kei133. Further investigations are required to determine whether these factors restrict the effect of overexpression of ELO3 and AUR1, or whether stronger overexpression of the genes would result in changes in the lipid profile.
ORM1/2 deletion increases ceramide levels but reduces the fraction of complex sphingolipids
The SPT complex, catalysing the formation of LCB from C16 and C18 fatty acids, is a rate-limiting enzyme complex in sphingolipid production15 and thus likely exerts a high flux control. Overexpression of the genes encoding enzymes in the complex will probably have a limited effect on LCB production, as they are suppressed by the negative regulators ORM1 and ORM215. Therefore, ORM1 and ORM2 were deleted in an attempt to increase the flux through the sphingolipid pathway. The resulting orm1/2Δ mutant exhibited a significantly impaired viability in aerobic shake flasks with buffered minimal medium, so a richer medium supplemented with amino acids was used to reduce the metabolic burden. The strain was cultivated in pH- and oxygen-controlled bioreactors, thereby avoiding the variation in aeration and temperature that normally occurs when shake flasks are removed from the incubator for sampling. This set-up improved the cell viability of the orm1/2Δ mutants to 89% at mid-exponential growth, while the wild type remained fully viable (Table 3). Another reason for adding amino acids to the minimal medium was to ensure that serine shortage did not limit sphingolipid production, as the SPT complex has been shown to be regulated upon serine availability22,34.
Physiological characterization of aerobic batch bioreactor cultivations revealed a 19% lower maximum specific growth rate, a 30% lower ethanol yield and similar biomass yield on glucose, when comparing the orm1/2Δ mutant to the wild type. A decrease in ethanol yield, as observed here, is usually associated with an increase in biomass yield due to elevated respiration. However, this was not observed in the orm1/2Δ mutant due to biomass loss by the lysis of non-viable cells, as indicated by the lower final biomass concentration after all the carbon sources had been depleted, and the difficulties to close the carbon balance for this specific mutant. The glycerol yield was 46% higher than in the wild type, which is a typical sign of increased cellular stress35.
Lipid analysis showed a decrease in all three complex sphingolipid classes (IPC, MIPC and MIP2C), although these were only statistically significant for IPC and MIP2C, which decreased by 82% and 61%, respectively, in the orm1/2Δ mutant compared to the wild type (Fig. 2a). The opposite trend was observed for the precursor ceramide, which increased on average by 680% in the orm1/2Δ mutant compared to the wild type (Fig. 2b). In addition, the glycerophospholipid class PI, required for the synthesis of complex sphingolipids decreased by 63% in the orm1/2Δ mutant compared with the wildtype, while only minor differences were observed for the other glycerophospholipid classes (Fig. 2c). Owing to the lower degree of unsaturation of PI compared to other glycerophospholipid classes, the decrease in PI also contributed to an increase in the total glycerophospholipid unsaturation level (Fig. 2d). Fatty acyl chain length increased slightly in the complex sphingolipids and ceramides, and decreased slightly in the total glycerophospholipids (Fig. 2e,f,g).
The results of previous studies on the effect of the orm1/2Δ on the lipid profile are inconsistent. Some studies have reported an increase in the ceramide content15,36, as in the present study, while others reported a decrease37. Furthermore, the total level of complex sphingolipids has been reported to increase15 as well as decrease36. To the best of our knowledge, no previous study has described the effect of the orm1/2Δ on the glycerophospholipid profile. Serine, used for LCB synthesis, and inositol, used for PI synthesis, are critical medium components affecting sphingolipid synthesis22,37, and a difference in medium composition could perhaps explain the discrepancy in the results reported so far. Breslow et al.15 used a complex YPD medium, Han et al.37 used a minimal medium with 36 g/L myo-inositol, Shimobayashi et al.36 used a minimal medium with 2 mg/L myo-inositol, while in the present study a minimal medium supplemented with amino acids, including 1.1 g/L serine and 25 mg/L myo-inositol, was used.
The increase in ceramide seen in the orm1/2Δ mutant, compared to the wildtype, in the present study suggests that the flux control has shifted from the SPT complex to Aur1. In addition, the decrease in complex sphingolipids indicates not only a shift of the rate controlling step but a reduced rate of the Aur1 reaction, perhaps due to low levels of PI, the second substrate of Aur1. In fact, lipid analysis showed reduced total levels of PI in the cell, although the PI concentrations in the Golgi, used by Aur1, may not mirror the total levels of PI. Instead the decrease in complex sphingolipids could be linked to a described second function of the Orm proteins in stimulating the synthesis of complex sphingolipids in the Golgi, possibly through the activation of Sac136. Sac1 cycles from the ER, where it interacts physically with the Orm proteins, to the Golgi, where it catalyses the release of PI, required for the synthesis of complex sphingolipids by Aur138. Perhaps the absence of Orm proteins leads to a dysfunctional Sac1, not capable of catalysing the release of PI in the Golgi. This hypothesis is supported by the fact that deletion of the SAC1 gene significantly decreases the levels of both PI and complex sphingolipids38. Alternatively, the low PI and complex sphingolipid levels suggest inositol deficiency, as inositol is required for PI synthesis and the orm1/2Δ mutant has previously been shown to be an inositol auxotroph37. However, our medium contained 25 mg/L myo-inositol, which should be sufficient as the nutrient levels in this medium have been set so as to not limit growth to when using 2% glucose as carbon source21. Furthermore, commercial mineral medium (YNB, MP Biomedicals) contains only 2 mg/L myo-inositol.
ELO3 and AUR1 overexpression in combination with ORM1/2 deletion partly reverses the growth defect of the ORM1/2 deletion mutant, but it does not increase the fraction of complex sphingolipids
Only a combined design, targeting the enzymes of the major bottlenecks in the sphingolipid biosynthesis pathway, will fully reveal the effect of the engineering strategy. The ELO3-AUR1-OE mutant exhibited no significant physiological or lipid profile differences compared to the wild type, although additional effects were observed in combination with orm1/2Δ. The low viability and specific growth rate of the orm1/2Δ mutant was substantially improved in the ELO3-AUR1-OE-orm1/2Δ mutant, reaching levels similar to those in the wild type (Table 3). This beneficial effect of ELO3-AUR1-OE on the orm1/2Δ mutant is in line with an observation from a large genetic interaction screen showing that AUR1 was the gene among 1400 damped or deleted genes that exacerbated the effect of orm2Δ most15, as previously discussed36. In addition, the average ceramide level was lower in the ELO3-AUR1-OE-orm1/2Δ strain than in the orm1/2Δ mutant (Fig. 2b), suggesting an increase in downstream flux capacity due to AUR1 overexpression, potentially explaining the increased viability of this strain, as ceramides are known inducers of apoptosis39. The difference in ceramide levels was, however, not statistically significant, and additional studies must be performed before any conclusions can be drawn. Interestingly, no other major differences were observed in the lipid classes between the two strains (Fig. 2c), indicating that Aur1 despite being over expressed, still exerts the highest flux control of the pathway. The shortage in PI supply capacity due to the orm1/2Δ may prevent an increase in flux in the sphingolipid pathway in the ELO3-AUR1-OE-orm1/2Δ mutant.
When investigating the sphingolipid and glycerophospholipid chain length in detail, the ELO3-AUR1-OE-orm1/2Δ mutant showed an increase in both complex sphingolipid and glycerophospholipid chain length compared to the orm1/2Δ mutant – a difference which was not observed when comparing ELO3-AUR1-OE with the wild type (Fig. 2e,g). The increase in the sphingolipid classes was observed specifically in the percentage of C46 species present. The ceramide chain length profile showed the same trend, but this was not statistically significant (Fig. 2f). Sphingolipid chains with 44 carbons are most likely a combination of a C18 LCB and a C26 fatty acid, as this combination has previously been shown to be the most common sphingolipid species30. Synthesis of fatty acids longer than C26 have not been reported. Sphingolipid chains with 46 carbons should hence correspond to C20/C26 species, suggesting a fatty acid shift in the ELO3-AUR1-OE-orm1/2Δ mutant from C16 to C18, which is the fatty acid that gives rise to C20 LCB. This observed shift in fatty acids may thus be the effect of increased elongation in the fatty acid elongation complex due to the overexpression of ELO3.
General discussion
In our previous studies, on the membrane properties of Z. bailii8,12, we suggested the potential of membrane engineering as a tool to create more robust microbial cell factories. Engineering the cell to achieve specific changes in the lipid composition of the membrane may, however, prove to be a challenging task. The aim of the present study was to increase the fraction of complex sphingolipids in the cell membrane of S. cerevisiae by altering the expression of genes associated with the production of LCBs and VLCFAs, and the conversion of ceramide into complex sphingolipids. Approaches such as this, where several components of the sphingolipid metabolism are targeted simultaneously, have so far been rare. Most previous knowledge regarding sphingolipid metabolism has been obtained through investigations of a single step in the pathway or in large genetic screens40 with the aim of understanding genetic interactions, rather than changing the lipid profile. In the present study, we were able to demonstrate that combining ELO3-AUR1-OE with orm1/2Δ reduces the stress caused by the orm1/2Δ leading to improved cell growth and fermentative performance, as well as changes in fatty acyl chain length. However, the strategy did not result in an increase in complex sphingolipids, and can therefore not be used as a means of increasing microbial tolerance to inhibitory compounds. Importantly, we were able to gain new insights into the sphingolipid metabolism of S. cerevisiae that could aid in the design of new and more efficient engineering strategies.
The flux of a metabolic pathway is dependent on the amount and activity of the enzymes catalysing each step and the available metabolite concentrations. Difficulties may be encountered in strategies to increase the amount of enzymes available to catalyse a specific step on a genetic level, as in the present study, due to transcriptional regulation altering mRNA levels and post-translational regulation altering enzyme activity. Sphingolipid biosynthesis has recently been shown to be strongly regulated by phosphorylation41, so a strategy targeting regulatory enzymes instead of structural enzymes may be more favourable, although such a strategy would be difficult to design due to our, as yet, incomplete understanding of regulation of the sphingolipid metabolism42. The double role played by sphingolipids in the cell, as structural building blocks in the cell membrane, and as signalling molecules involved in many cellular functions, further complicates the picture42. The main role of some enzymes is to modify sphingolipid concentrations for signalling purposes, and these may not be involved in regulating the bulk concentration of complex sphingolipids, as they are located in different compartments43. Furthermore, a comprehensive design targeting all the critical steps in the pathway is required to ensure that substrate is available for each of the individual biosynthetic steps. In sphingolipid metabolism, it is also essential to maintain the correct balance between pathway intermediates such as VLCFAs, LCBs and ceramides to avoid the toxic effects of these intermediates at elevated levels44. In mammalian cells, the ceramide transport protein (CERT) is essential to relieve the stress induced by high ceramide levels45. CERT transports ceramides produced in the ER to the Golgi, where they are used for the production of complex sphingolipids. A similar function was recently found in yeast by Prinz and co-workers, who discovered that Nvj2 had a ceramide transfer function which, when overexpressed, relieved the ceramide-induced stress in S. cerevisiae46. For future engineering strategies we suggest the combination of NVJ2 overexpression and AUR1 overexpression to ensure that ceramides in the ER are available in the Golgi for use by Aur1 to synthesize complex sphingolipids. Finally, membrane sphingolipids can be in fact considered as a metabolic intermediate rather than a final product, as they are recycled by the cell47. Therefore, sphingolipid recycling reactions can be also considered as a potential target.
Difficulties in increasing pathway flux due to post-translational regulation and keeping toxic pathway intermediates sufficiently low, led us to suggest an alternative strategy, namely one aimed at introducing a pathway bottleneck elsewhere in lipid metabolism. This strategy relies on the fact that the cell itself redirects the flux to increase the desired group of lipids, in order to synthesize sufficient levels of lipids to maintain a functional plasma membrane. This strategy may seem farfetched, but reducing the flux not only limits the problems associated with unexpected post-translation modification, but it is also significantly more straightforward to construct a single rate-controlling step than to achieve increased flux through the entire metabolic pathway. For example, enzymes catalysing glycerophospholipid production could be downregulated to increase the fraction of complex sphingolipids in the cell, mimicking the response to an inhibitory concentration of acetic acid observed in Z. bailii, where a considerable decrease in glycerophospholipid was compensated by a high increase in complex sphingolipids12.
To prove the value of membrane engineering, an interesting alternative to modification of the native lipid production pathways of the cells could be membrane alteration using heterologous lipids, thereby circumventing cellular regulation. Archaea lipids and their properties have been studied extensively, as reviewed by Matsumi et al.48, and the introduction of trans fatty acids into E. coli has recently been shown to successfully increase its tolerance to many alcohols, aromatic solvents and organic acids11.
Conclusions
In an attempt to increase the amount of complex sphingolipids in S. cerevisiae, ELO3 overexpression, AUR1 overexpression and ORM1/2 deletion were performed. Neither ELO3 nor AUR1 overexpression influenced the lipid metabolism in our set-up, while orm1/2Δ instead caused a decrease in both complex sphingolipids and PI glycerophospholipids. However, by combining ELO3-AUR1-OE with the orm1/2Δ, the ELO3-AUR1-OE background alleviated the reduction in growth caused by the orm1/2Δ, and increased the fatty acyl chain length, demonstrating that overall approaches combining alterations in several steps in the pathway may give additional effects not observed with the single mutations. We suggest that our findings can be used as the foundation for further studies on sphingolipid metabolism. Furthermore, to begin evaluating the potential of membrane engineering, we recommend that changes are made in the cell membrane that can be achieved by altering only a few genes, or the introduction of new lipid species by the expression of genes encoding heterologous synthesis pathways in order to avoid cellular regulation.
Additional Information
How to cite this article: Lindahl, L. et al. Membrane engineering of S. cerevisiae targeting sphingolipid metabolism. Sci. Rep. 7, 41868; doi: 10.1038/srep41868 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Committee on Industrialization of Biology: A Roadmap to Accelerate the Advanced Manufacturing of Chemicals; Board on Chemical Sciences and Technology; Board on Life Sciences; Division on Earth and Life Studies; National Research Council. Industrialization of Biology: A Roadmap to Accelerate the Advanced Manufacturing of Chemicals. (Washington (DC): National Academies Press: (US), (29 Jun 2015).
Zhu, L., Zhu, Y., Zhang, Y. & Li, Y. Engineering the robustness of industrial microbes through synthetic biology. Trends in Microbiology 20, 94–101, doi: 10.1016/j.tim.2011.12.003 (2012).
Dunlop, M. J. Engineering microbes for tolerance to next-generation biofuels. Biotechnology for Biofuels 4, doi: 10.1186/1754-6834-4-32 (2011).
Nicolaou, S. A., Gaida, S. M. & Papoutsakis, E. T. A comparative view of metabolite and substrate stress and tolerance in microbial bioprocessing: From biofuels and chemicals, to biocatalysis and bioremediation. Metabolic Engineering 12, 307–331, doi: 10.1016/j.ymben.2010.03.004 (2010).
Huffer, S., Clark, M. E., Ning, J. C., Blanch, H. W. & Clark, D. S. Role of alcohols in growth, lipid composition, and membrane fluidity of yeasts, bacteria, and archaea. Applied and Environmental Microbiology 77, 6400–6408, doi: 10.1128/AEM.00694-11 (2011).
Liu, S. & Qureshi, N. How microbes tolerate ethanol and butanol. New Biotechnology 26, 117–121, doi: 10.1016/j.nbt.2009.06.984 (2010).
Warth, A. D. Transport of benzoic and propanoic acids by Zygosaccharomyces bailii . Journal of General Microbiology 135, 1383–1390 (1989).
Lindahl, L., Genheden, S., Eriksson, L. A., Olsson, L. & Bettiga, M. Sphingolipids contribute to acetic acid resistance in Zygosaccharomyces bailii . Biotechnology and Bioengineering 113, 744–753, doi: 10.1002/bit.25845 (2016).
Yazawa, H., Kamisaka, Y., Kimura, K., Yamaoka, M. & Uemura, H. Efficient accumulation of oleic acid in Saccharomyces cerevisiae caused by expression of rat elongase 2 gene (rELO2) and its contribution to tolerance to alcohols. Applied Microbiology and Biotechnology 91, 1593–1600, doi: 10.1007/s00253-011-3410-4 (2011).
Zheng, D. Q. et al. Comparative functional genomics to reveal the molecular basis of phenotypic diversities and guide the genetic breeding of industrial yeast strains. Applied Microbiology and Biotechnology 97, 2067–2076, doi: 10.1007/s00253-013-4698-z (2013).
Tan, Z., Yoon, J. M., Nielsen, D. R., Shanks, J. V. & Jarboe, L. R. Membrane engineering via trans unsaturated fatty acids production improves Escherichia coli robustness and production of biorenewables. Metabolic Engineering 35, 105–113, doi: 10.1016/j.ymben.2016.02.004 (2016).
Lindberg, L., Santos, A. X. S., Riezman, H., Olsson, L. & Bettiga, M. Lipidomic Profiling of Saccharomyces cerevisiae and Zygosaccharomyces bailii Reveals Critical Changes in Lipid Composition in Response to Acetic Acid Stress. PLoS ONE 8, e73936, doi: 10.1371/journal.pone.0073936 (2013).
Pinto, W. J., Wells, G. W. & Lester, R. L. Characterization of enzymatic synthesis of sphingolipid long-chain bases in Saccharomyces cerevisiae: Mutant strains exhibiting long-chain-base auxotrophy are deficient in serine palmitoyltransferase activity. Journal of Bacteriology 174, 2575–2581 (1992).
Gable, K., Slife, H., Bacikova, D., Monaghan, E. & Dunn, T. M. Tsc3p is an 80-amino acid protein associated with serine palmitoyltransferase and required for optimal enzyme activity. Journal of Biological Chemistry 275, 7597–7603, doi: 10.1074/jbc.275.11.7597 (2000).
Breslow, D. K. et al. Orm family proteins mediate sphingolipid homeostasis. Nature 463, 1048–1053 (2010).
Tehlivets, O., Scheuringer, K. & Kohlwein, S. D. Fatty acid synthesis and elongation in yeast. Biochimica et Biophysica Acta - Molecular and Cell Biology of Lipids 1771, 255–270, doi: 10.1016/j.bbalip.2006.07.004 (2007).
Dickson, R. C. New insights into sphingolipid metabolism and function in budding yeast. Journal of Lipid Research 49, 909–921, doi: 10.1194/jlr.R800003-JLR200 (2008).
Cowart, L. A. & Obeid, L. M. Yeast sphingolipids: Recent developments in understanding biosynthesis, regulation, and function. Biochimica et Biophysica Acta - Molecular and Cell Biology of Lipids 1771, 421–431, doi: 10.1016/j.bbalip.2006.08.005 (2007).
Entian, K. D. & Kötter, P. Yeast Genetic Strain and Plasmid Collections. In Methods in Microbiology Vol. 36, 629–666 (2007).
Amberg, D. C., Burke, D. & Strathern, J. N. Methods in Yeast Genetics: A Cold Spring Harbor Laboratory Course Manual. (Cold Spring Harbor Laboratory Press, 2005).
Verduyn, C., Postma, E., Scheffers, W. A. & Van Dijken, J. P. Effect of benzoic acid on metabolic fluxes in yeasts: A continuous-culture study on the regulation of respiration and alcoholic fermentation. Yeast 8, 501–517, doi: 10.1002/yea.320080703 (1992).
Cowart, L. A. & Hannun, Y. A. Selective substrate supply in the regulation of yeast de novo sphingolipid synthesis. Journal of Biological Chemistry 282, 12330–12340, doi: 10.1074/jbc.M700685200 (2007).
Koressaar, T. & Remm, M. Enhancements and modifications of primer design program Primer3. Bioinformatics 23, 1289–1291, doi: 10.1093/bioinformatics/btm091 (2007).
Untergasser, A. et al. Primer3-new capabilities and interfaces. Nucleic Acids Research 40, doi: 10.1093/nar/gks596 (2012).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408, doi: 10.1006/meth.2001.1262 (2001).
Guan, X. L., Riezman, I., Wenk, M. R. & Riezman, H. Yeast Lipid Analysis and Quantification by Mass Spectrometry. In Methods in Enzymology Vol. 470, 369–391 (Academic Press, 2010).
Choi, J. Y. & Martin, C. E. The Saccharomyces cerevisiae FAT1 gene encodes an acyl-coA synthetase that is required for maintenance of very long chain fatty acid levels. Journal of Biological Chemistry 274, 4671–4683, doi: 10.1074/jbc.274.8.4671 (1999).
Tani, M. & Kuge, O. Defect of synthesis of very long-chain fatty acids confers resistance to growth inhibition by inositol phosphorylceramide synthase repression in yeast Saccharomyces cerevisiae . Journal of Biochemistry 148, 565–571, doi: 10.1093/jb/mvq090 (2010).
Zimmermann, C. et al. TORC1 Inhibits GSK3-Mediated Elo2 Phosphorylation to Regulate Very Long Chain Fatty Acid Synthesis and Autophagy. Cell Reports 5, 1036–1046, doi: 10.1016/j.celrep.2013.10.024 (2013).
Ejsing, C. S. et al. Global analysis of the yeast lipidome by quantitative shotgun mass spectrometry. Proceedings of the National Academy of Sciences of the United States of America 106, 2136–2141, doi: 10.1073/pnas.0811700106 (2009).
Tabuchi, M., Audhya, A., Parsons, A. B., Boone, C. & Emr, S. D. The phosphatidylinositol 4,5-biphosphate and TORC2 binding proteins Slm1 and Slm2 function in sphingolipid regulation. Molecular and Cellular Biology 26, 5861–5875, doi: 10.1128/MCB.02403-05 (2006).
Denic, V. & Weissman, J. S. A Molecular Caliper Mechanism for Determining Very Long-Chain Fatty Acid Length. Cell 130, 663–677, doi: 10.1016/j.cell.2007.06.031 (2007).
Sato, K., Noda, Y. & Yoda, K. Kei1: A novel subunit of inositolphosphorylceramide synthase, essential for its enzyme activity and golgi localization. Molecular Biology of the Cell 20, 4444–4457, doi: 10.1091/mbc.E09-03-0235 (2009).
Montefusco, D. J. et al. Sphingoid bases and the serine catabolic enzyme CHA1 define a novel feedforward/feedback mechanism in the response to serine availability. Journal of Biological Chemistry 287, 9280–9289, doi: 10.1074/jbc.M111.313445 (2012).
Gasch, A. P. The environmental stress response: a common yeast response to diverse environmental stresses. In Yeast Stress Responses 11–70 (Springer Berlin Heidelberg, 2003).
Shimobayashi, M., Oppliger, W., Moes, S., Jenö, P. & Hall, M. N. TORC1-regulated protein kinase Npr1 phosphorylates Orm to stimulate complex sphingolipid synthesis. Molecular Biology of the Cell 24, 870–881 (2013).
Han, S., Lone, M. A., Schneiter, R. & Chang, A. Orm1 and Orm2 are conserved endoplasmic reticulum membrane proteins regulating lipid homeostasis and protein quality control. Proceedings of the National Academy of Sciences of the United States of America 107, 5851–5856, doi: 10.1073/pnas.0911617107 (2010).
Brice, S. E., Alford, C. W. & Cowart, L. A. Modulation of sphingolipid metabolism by the phosphatidylinositol-4-phosphate phosphatase Sac1p through regulation of phosphatidylinositol in Saccharomyces cerevisiae. Journal of Biological Chemistry 284, 7588–7596, doi: 10.1074/jbc.M808325200 (2009).
Rego, A. et al. The yeast model system as a tool towards the understanding of apoptosis regulation by sphingolipids. FEMS Yeast Research 14, 160–178, doi: 10.1111/1567-1364.12096 (2014).
Dickson, R. C. Roles for Sphingolipids. In Saccharomyces cerevisiae in Sphingolipids as Signaling and Regulatory Molecules 217–231 (Springer New York, 2010).
Da Silveira Dos Santos, A. X. et al. Systematic lipidomic analysis of yeast protein kinase and phosphatase mutants reveals novel insights into regulation of lipid homeostasis. Molecular Biology of the Cell 25, 3234–3246, doi: 10.1091/mbc.E14-03-0851 (2014).
Aguilera-Romero, A., Gehin, C. & Riezman, H. Sphingolipid homeostasis in the web of metabolic routes. Biochimica et Biophysica Acta - Molecular and Cell Biology of Lipids 1841, 647–656, doi: 10.1016/j.bbalip.2013.10.014 (2014).
Hannun, Y. A. & Obeid, L. M. The ceramide-centric universe of lipid-mediated cell regulation: Stress encounters of the lipid kind. Journal of Biological Chemistry 277, 25847–25850, doi: 10.1074/jbc.R200008200 (2002).
Kobayashi, S. D. & Nagiec, M. M. Ceramide/long-chain base phosphate rheostat in Saccharomyces cerevisiae: Regulation of ceramide synthesis by Elo3p and Cka2p. Eukaryotic Cell 2, 284–294, doi: 10.1128/EC.2.2.284-294.2003 (2003).
Hanada, K., Kumagai, K., Tomishige, N. & Yamaji, T. CERT-mediated trafficking of ceramide. Biochimica et Biophysica Acta - Molecular and Cell Biology of Lipids 1791, 684–691, doi: 10.1016/j.bbalip.2009.01.006 (2009).
LIU, L.-K., Toulmay, A. & Prinz, W. An ER-Golgi tether facilitates ceramide transfer out of the ER and alleviates ceramide toxicity. The FASEB Journal 30, 660.661 (2016).
Klug, L. & Daum, G. Yeast lipid metabolism at a glance. FEMS Yeast Research 14, 369–388, doi: 10.1111/1567-1364.12141 (2014).
Matsumi, R., Atomi, H., Driessen, A. J. M. & van der Oost, J. Isoprenoid biosynthesis in Archaea - Biochemical and evolutionary implications. Research in Microbiology 162, 39–52, doi: 10.1016/j.resmic.2010.10.003 (2011).
Acknowledgements
We are grateful to Prof. Howard Riezman for fruitful discussions, and allowing us to use his equipment for lipid analysis. Financial support from the Swedish Energy Agency, the Swedish Research Council, the Chalmers Energy Area of Advance, the Swiss National Science Foundation, and the Swiss Initiative in Systems Biology, ‘SystemsX.ch’ is gratefully acknowledged.
Author information
Authors and Affiliations
Contributions
All the authors were involved in conceiving and designing the experiments, and the analysis of the data. L.L., A.X.S.S. and H.O. performed the experiments and L.L. and H.O. wrote the paper. All the authors read and approved the final paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Lindahl, L., Santos, A., Olsson, H. et al. Membrane engineering of S. cerevisiae targeting sphingolipid metabolism. Sci Rep 7, 41868 (2017). https://doi.org/10.1038/srep41868
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep41868
This article is cited by
-
Studying on genetic diversity and metabolic differences of Saccharomyces cerevisiae in Baijiu
European Food Research and Technology (2024)
-
Analysis of the response of the cell membrane of Saccharomyces cerevisiae during the detoxification of common lignocellulosic inhibitors
Scientific Reports (2021)
-
Genome-wide search for candidate genes for yeast robustness improvement against formic acid reveals novel susceptibility (Trk1 and positive regulators) and resistance (Haa1-regulon) determinants
Biotechnology for Biofuels (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
