
Abstract
The CRISPR/Cas9 system is a recently developed genome editing technique. In this study, we used a modified CRISPR system, which employs the fusion of inactive Cas9 (dCas9) and the FokI endonuclease (FokI-dCas9) to correct the most common variant (allele frequency 21.4%) in the phenylalanine hydroxylase (PAH) gene - c.1222C>T (p.Arg408Trp) - as an approach toward curing phenylketonuria (PKU). PKU is the most common inherited diseases in amino acid metabolism. It leads to severe neurological and neuropsychological symptoms if untreated or late diagnosed. Correction of the disease-causing variants could rescue residual PAH activity and restore normal function. Co-expression of a single guide RNA plasmid, a FokI-dCas9-zsGreen1 plasmid, and the presence of a single-stranded oligodeoxynucleotide in PAH_c.1222C>T COS-7 cells – an in vitro model for PKU – corrected the PAH variant and restored PAH activity. Also in this system, the HDR enhancer RS-1 improved correction efficiency. This proof-of-concept indicates the potential of the FokI-dCas9 system for precision medicine, in particular for targeting PKU and other monogenic metabolic diseases.
Similar content being viewed by others

Introduction
Phenylketonuria (PKU, OMIM 261600) is the most frequent and important inherited metabolic disease, caused by deficiency of hepatic phenylalanine hydroxylase (PAH, EC 1.14.16.1). PAH converts phenylalanine (Phe) to tyrosine (Tyr). The cofactor tetrahydrobiopterin (BH4), iron and molecular oxygen are required for this process1. PAH deficiency results in the accumulation of Phe in the body including the brain. If untreated or diagnosed late, its high concentration leads to severe neurological and neuropsychological symptoms, including intellectual disability, seizures, ataxia, motor deficits and behavioral problems. Because of the severity of the disease, newborn screening for PKU has been instituted in many countries. Lifelong restriction of dietary Phe is the established treatment for preventing neuronal damage. However, poor compliance by PKU patients and the heavy economic burden challenge the dietary management. Although the restriction of dietary Phe improves cognitive performance, other neuropsychological parameters such as choice reaction time, attention, and executive functions are impaired compared to healthy controls even in the best treated individuals. An additional supplement of tetrahydrobiopterin (BH4; sapropterin dihydrochloride) may be beneficial for some PKU patients with a mild pheotype2. However, the ideal approach for curing PKU would be to correct variants in the PAH gene directly through gene therapy. Since PKU is an autosomal recessive inherited disorder, only individuals with PAH variants located in both alleles develop symptoms. Even if only one allele is corrected, the residual PAH activity would be sufficiently increased, abolishing or reducing PKU symptoms. In patients with severe classic PKU, the c.1222C>T (p.Arg408Trp) single nucleotide variant is the most frequent one in PAH (allele frequency 21.4%) and p.[Arg408Trp];[Arg408Trp] the most common genotype (genotype frequency 10.2%)3. Thus, PAH_c.1222C>T was chosen as the target in this study.
The most promising genetic repair of variants is achievable through homologous recombination repair (homology directed repair; HDR) after the introduction of DNA double-stranded breaks (DSBs) close to the variant. DSBs can be generated by different genome editing technologies, such as zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs) and the recently developed technique of clustered regularly interspaced short palindromic repeats (CRISPR)4. The CRISPR/Cas9 system was first identified as a prokaryotic defense mechanism against bacteriophages, and is currently being used for site-specific genome editing. It is composed of a single-guide RNA (sgRNA) containing a 20 nt sequence that targets a specific genomic site and one Cas9 nuclease that enables the cleavage of genomic DNA. The DSBs then stimulate the DNA repair pathway through non-homologous end joining (NHEJ) and HDR. Of these mechanisms, HDR can generate the desired sequence replacement at the DSBs through the usage of a donor DNA template, which corrects the specific variant within the genome5. Several studies have recently reported encouraging results using CRISPR/Cas9 for successful therapeutic concepts, such as Duchenne muscular dystrophy and retinitis pigmentosa in vitro and in vivo6,7,8,9,10. However, there are still two major challenges in the CRISPR/Cas9-mediated genome correction: frequent off-target effects and low HDR efficiency5,11,12. To overcome these problems, different strategies were adopted in this study. First, we chose the CRISPR RNA-guided FokI nuclease system (FokI-dCas9 system)13,14,15 instead of the CRISPR/Cas9 system as the specific genome editing tool. The FokI-dCas9 system is composed of two distinct sgRNAs and FokI-dCas9 nuclease, which is a fusion of inactive Cas9 (dCas9) and the FokI endonuclease. The FokI-dCas9 system differs from CRISPR/Cas9 in that the simultaneous binding of two distinct FokI-dCas9:sgRNAs is required. Moreover, we employed 3-(N-benzylsulfamoyl)-4-97 bromo-N-(4-bromophenyl)benzamide (RS-1), an HDR enhancer16,17, as a RAD51-stimulatory compound18. By this means, we successfully performed a genetic repair of the c.1222C>T PAH variant, which for the first time corrected the point variant and rescued both PAH protein expression and activity.
Results
Establishment of the PAH_c.1222C>T COS-7 cell line as an in vitro PKU model
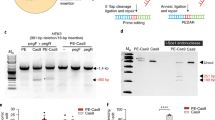
The PAH_c.1222C>T variant was introduced into COS-7 cells by transfection with an appropriately modified vector DNA. Single clones were sub-cultured for at least 15 passages. Sanger sequencing analysis proved that the “T” point variant was appropriately located in PAH (Fig. 1a). Reverse transcription PCR (RT-PCR) confirmed that PAH_c.1222C>T was expressed in the transfected COS-7 cells at the RNA level at a similar level as the wild-type protein used in control; untransfected COS-7 cells exhibited no PAH RNA (Fig. 1b). Concurrently, Western blot analysis with an antibody against the wild-type protein (PAH_WT) revealed a basically complete loss of the PAH protein in the cells transfected with the mutated gene version (Fig. 1c).

PAH_c.1222C>T COS-7 cell line was established as a PKU in vitro model.
(a) Sanger sequencing analysis of PAH_c.1222C>T COS-7 cells by IRES reverse primer showed “A” point mutation on the PAH gene. (b) RT-PCR confirmed PAH_c.1222C>T expression in established cell line. (c) Western blot showed that the expression of PAH in PAH_c.1222C>T cells was lower than in PAH_WT cells.
The PAH_c.1222C>T variant was corrected using the FokI-dCas9 system
The FokI-dCas9 system is different from the CRISPR/Cas9 system insofar as only binding of a FokI-dCas9 dimer can cleave DNA (Fig. 2a). After electroporation of COS-7 cells with expression vectors of the FokI-dCas9 gene and the sgRNA, flow cytometry analysis showed that approximately 18.4% of cells co-expressed them simultaneously (Fig. 2b). In presence of RS-1, PAH expression in PAH_c.1222C>T COS-7 cells treated with 0.5 or 1 nM of the single-stranded oligodeoxynucleotide (ssODN) that represents the replacement sequence was significantly higher than the PAH expression of PAH_c.1222C>T COS-7 cells that were not transfected with the vectors. A lower ssODN concentration had no apparent effect on PAH expression (Fig. 2c). Furthermore, liquid chromatography-electrospray ionization tandem mass spectrometry was employed to evaluate the PAH activity after repair (Fig. 2d), confirming the results of the Western blot analysis. Sanger sequencing analysis was performed to confirm the correction in all groups. The 1 nM ssODN and 0.5 nM ssODN groups showed double-peaks at the corrected position regardless of RS-1 treatment, demonstrating the presence of both a “T” and a “C” at the PAH_c.1222C>T variant in the cell population (Supplementary Fig. 1a). To examine the correction rate, we amplified by PCR target DNA from the 1 nM ssODN with RS-1 treatment group and verified the sequence by TA-cloning. Sequence analysis of the DNA of 30 colonies showed that 8 of them (26.7%) contained the correct “C” (Fig. 2e). To assess the possible off-targeting introduced by FokI-dCas9-directed cleavage, two potential off-target sites were predicted by CasOT (Supplementary Table 1). Both potential sites were analyzed together with the PAH gene on-target site by Sanger sequencing. None of the analyzed regions showed evidence of off-target cleavage (data not shown).

Correction of the PAH_c.1222C>T variant in COS-7 cells.
(a) Scheme of the process: one dimer of the FokI-dCas9 complex binds to two “half-sites” on the genome with a certain spacer length and generate DSBs. DSBs are then repaired by HDR. (b) Co-expression efficiency of the FokI-dCas9 and sgRNA plasmids in PAH_c.1222C>T COS-7 cells was analyzed by flow cytometry. (c) The PAH expression of each cell population was detected by Western blot. (d) The PAH activity of each group was measured by liquid chromatography-electrospray ionization tandem mass spectrometry. (e) Correction of variant; one of 30 sequences are shown. T/A-cloning into E. coli and Sanger sequencing were performed on DNA isolated from individual COS-7 cells transfected with the FokI-dCas9 and sgRNA plasmids as well as 1 nM ssODN and treated with RS-1. Among all 30 randomly picked colonies, representing the DNA of 30 COS-7 cells, 26.7% of the cells exhibited correction of the variant.
Discussion
Although the restriction of dietary Phe and/or supplementation with BH4 can substantially alleviate clinical symptoms of PKU, patients do not manage to maintain dietary treatment for life. They suffer from impaired neuropsychological and partly from intellectual function and PKU women are at risk for the maternal PKU syndrome. The ideal approach to treating this disease would be a correction of the PAH variant. PKU gene therapy has been developed over the past two decades using an animal model. Researchers delivered the wild-type PAH gene to tissues with an adeno-associated virus (AAV) vector. However, the temporary recovery of PAH activity, risks associated with the AAV vector or a very low gene transfer rate in the past limited the development of this treatment option19,20. Ongoing research is focused on new genome editing technologies that can correct genomic variants. Therefore, in this study the FokI-dCas9 system was chosen to correct with high accuracy and efficiency the most common PAH variant in cells.
The CRISPR/Cas9 system stimulates DNA repair mechanisms such as NHEJ and HDR. NHEJ leads to random deletion or insertion variants close to the DSB site, resulting in gene knock out, while HDR can generate a desired sequence replacement at the DSB by donating a DNA template, resulting in gene correction. Despite the tremendous potential of the original CRISPR/Cas9 system21, several challenges still limit its application to gene therapy. For example, the short 20 bp target sequence required in the sgRNA, of the commonly used SpCas9 system may cause off-target effects elsewhere in the genome12. Moreover, NHEJ is the dominant mechanism for repairing DSBs after DNA cleavage in mammals, preventing gene correction by HDR. Thus, new strategies that avoid off-target effects and increase the HDR:NHEJ ratio are needed22.
The FokI-dCas9 system can greatly improve the specificity of genome editing in vitro and in vivo. In contrast to the CRISPR/Cas9 system, it requires dimerization of the FokI-dCas9-sgRNA complex, meaning that monomeric FokI-dCas9-sgRNA is unable to cut the DNA strand14. Three rules are used in designing a specific FokI-dCas9-sgRNA complex that retains high cleavage efficiency. First, the pair of sgRNAs should bind to sense and antisense strands. Second, two distinct FokI-dCas9-sgRNA-binding “half-sites” must be located close to the targeted genomic position, and third, the FokI-dCas9-sgRNA complex requires a particular orientation. Generally, having protospacer adjacent motif (PAM) sequences for the two distinct FokI-dCas9-sgRNA complexes located on the outer boundaries of the target site (“PAM-out” orientation) is superior to having them connected directly to the spacer sequence (“PAM-in” orientation). In addition, the spacer length between two PAM sequences has limitations. Fourteen to 17 bp of spacer length exhibit higher on-target cleavage efficiency than other spacer lengths between 0 to 31 bp15. Based on these rules, this study chose the FokI-dCas9-sgRNA complex with a “PAM-out” orientation and spacer length of 17 bp. Moreover, it is worth noting that while a single FokI-dCas9-sgRNA can bind elsewhere in the genome, it cannot cleave DNA and cause off-target effects. In addition, FokI-dCas9 targets DNA sites with more than 140-fold higher specificity than normal SpCas9 and higher specificity than Cas9 nickases, which only cut single DNA strands throughout the genome23.
Although besides FokI-dCas9, strategies like Cas9 nickases or high-fidelity eSpCas9 variants24,25 can be adopted to enhance Cas9 specificity, we selected the FokI-dCas9 system to correct the PAH variant for its dual advantage of high specificity14 and potential improvement of HDR efficiency by DNA template preservation. It is generally known that the generation of HDR requires a large amount of DNA template. In the CRISPR/Cas9, eSpCas9 or Cas9 nickases systems, the DNA template can be cut by nucleases, even if an ssODN is used as the repair template. However, the FokI-dCas9-sgRNA complex cannot cleave the ssODN template as two distinct FokI-dCas9-sgRNA complexes have to bind to different DNA strands. Therefore, the CRISPR RNA-guided FokI nucleases system ensures a rich source of repair template during the HDR process. To further enhance HDR efficiency, a small-molecule drug was used in our study. The potent NHEJ inhibitor 5,6-bis((E)-benzylideneamino)-2- mercaptopyrimidin-4-ol (SCR-7) and the HDR enhancer RS-1, can be used to improve nuclease-mediated HDR efficiency16,26. RS-1 shows greater potential than SCR-7 in improving the HDR rate in vivo17. Our study found that, after electroporation with the FokI-dCas9-sgRNA complex, the PAH_c.1222C>T COS-7 cells remaining in RS-1 culture medium showed more than twice as much PAH activity as negative controls. Also, the PAH expression of the RS-1 group was significantly higher than cells grown without RS-1. The 26.7% correction rate can be expected to provide a clinically sufficient therapeutic effect, as normal development has been reported in patients with PAH activities as low as 8.7–34.5% of normal27. RS-1 can increase knock-in efficiency by two- to five-fold in both the TALEN- and Cas9-mediated knock-in systems17. We found a similar improvement in HDR efficiency in the FokI-dCas9 system.
Using LC-ESI-MS-MS, we could determine in PAH_c.1222C>T COS-7 cells electroporated with the sgRNA plasmid, FokI-dCas9 plasmid and 1 nM ssODN and cultured in RS-1 a PAH activity of 22.1% compared to 8.8% in the control group. The goal of this study was not only the genetic repair of the PAH variant, but also the rescue of PAH activity after correction. The rescue of PAH activity demonstrates that our strategy is a promising approach for PKU treatment.
In conclusion, this study indicates that the FokI-dCas9 system is a suitable, specific and effective genome editing technique for correcting a major PAH variant that causes PKU. This highlights its tremendous potential for the precise treatment of also other inherited metabolic diseases.
Methods
Construction of the PAH_c.1222C>T vector and establishment of the PAH_c.1222C>T COS-7 cell line
A wild-type PAH cDNA was cloned into the pLVX-EF1α-IRES-Puro vector (Clontech) using Xbal and BamHI cleavage (Thermo Fisher Scientific) after the amplification of cDNA from the pCMV-FLAG-PAH wild-type plasmid. The PAH_c.1222C>T variant in the PAH cDNA sequence was introduced by site-directed mutagenesis using the QuikChange II XL Site-Directed Mutagenesis Kit (Agilent Technologies) and confirmed by Sanger DNA sequencing (GATC Biotech). EF1α forward and IRES reverse sequencing primers (Thermo Fisher Scientific) were used to sequence the PAH_c.1222C>T variant.
COS-7 cells were cultured at 37 °C with 5% CO2 in Dulbecco’s Modified Eagle Medium (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. One day prior to transfection, 2 × 106 cells were seeded and cultured in one dish of 10 cm diameter. The transfection of one dish utilized 45 μL Fugene HD reagent (Promega) and 15 μg PAH_c.1222C>T plasmid or the plasmid with the wild-type sequence as a control. Forty-eight hours post transfection, the growth medium was replaced with complete growth medium containing 10 μg/mL puromycin as the selective agent. After the non-transfected COS-7 cells had died, a single PAH_c.1222C>T COS-7 clone was isolated using limiting dilution cloning technique performed in 96-well plates with 10 μg/mL puromycin remaining in the culture medium. The clone was characterized through RT-PCR, and expanded in 75 cm2 cell culture flasks.
Construction of the CRISPR RNA-guided FokI system plasmids and the HDR donor
The IRES-ZsGreen1 fragment of the pLVX-EF1α-IRES-ZsGreen1 vector (Clontech, Mountain View, USA), was used to construct by means of the Megaprimer PCR method28 the FokI-dCas9-IRES-ZsGreen1 plasmid (Supplementary Fig. 1b).
gRNAs, targeting the PAH_c.1222C>T variant were designed with the E-CRISP software tool (www.e-crisp.org)29. One suitable gRNA pair, which is composed of two separate gRNAs, was chosen as previously described15. gRNA pairs should be located on either side of the target in the PAM-out orientation and contain spacer of 14–17 bp in length. The final sgRNA expression fragment (Supplementary Table 2) was synthesized using gBlocks (Integrated DNA Technologies). It contains two separate U6 promoters, gRNAs and tracrRNAs, but is free of any 5′ modifications. The synthesized sgRNA fragment was cloned into the pRSI9 vector (Cellecta) using Gibson Assembly Cloning (New England Biolabs) resulting in the pRSI9-1222sgRNA plasmid (Supplementary Fig. 1c).
The ssODN was synthesized using an HDR template from Integrated DNA Technologies. The homology arm of the HDR template located on the 5′end of the PAH_c.1222C>T variant position is 71 nt and the arm on the 3′end is 111nt (Supplementary Table 2).
Electroporation of PAH_c.1222C>T COS-7 cells
Electroporation was performed according to the instructions provided with the Gene Pulse Xcell electroporation system (Bio-Rad). Prior to electroporation, PAH_c.1222C>T COS-7 cells were washed with PBS and resuspended in Gene Pulser electroporation buffer (Bio-Rad) at 2.5 × 106 cells/mL. Ten micrograms of pRSI9-1222sgRNA plasmid, 10 μg FokI-dCas9-zsGreen plasmid and 0.1 nM to 1 nM ssODN were added to the 0.4 cm cuvette with 400 μL cells and mixed gently. The optimized electroporation setting for PAH_c.1222C>T COS-7 cells was previously determined to be the following: pulse type (square wave), V (200 V), PL (15 msec) and cuvette (0.4 cm). After electroporation, 400 μL of the electroporated cells were transferred into 6-well plates. These electroporated cells were cultured in complete growth medium with or without 15 μM RS-1 (Sigma-Aldrich) at 37 °C for 96 hours.
Flow cytometry analysis
Ninety-six hours after electroporation, cell pellets were harvested by centrifugation at 200 g for 4 min and resuspended in PBS buffer to 0.5 × 106 cells/mL. Flow cytometry analysis was performed on a BD FACSVerse™ flow cytometer (BD Biosciences). PAH_c.1222C>T COS-7 cells containing the 1222sgRNA plasmid were identified as PE positive while cells transfected with the FokI-dCas9-zsGreen plasmid were FITC positive. When the 1222sgRNA and FokI-dCas9-zsGreen plasmids were co-expressed, the cell group was gated as both PE and FITC positive group. The data were acquired and analyzed with FLOWJO software (FLOWJO).
Measurement of PAH activity by liquid chromatography-electrospray ionization tandem mass spectrometry
The PAH activity of the cell lysates was measured using previously described methods30. In brief, 5 μL (containing 5–10 μg of total protein) of cell homogenate obtained from PAH_c.1222C>T COS-7 cells after electroporation was mixed with 0.1 M Na-HEPES buffer, 2 μg catalase and 1 M L-Phe and incubated for 5 min. One μM Fe(NH4)2(SO4)2 was added into the mixture for one minute. PAH assay reaction was started by adding 200 μM BH4 and then incubated at 25 °C for 15 min. The reaction was stopped by adding 50 μL of 2% (w/v) acetic acid in ethanol. Then, the PAH samples were prepared for liquid chromatography-electrospray ionization tandem mass spectrometry in accordance with the EZ:faastTM kit manual (Phenomenex). Ten microliters of 100 μM Phe-d5 and 10 μL of 20 μM Tyr-d4 were added to 80 μL PAH assay samples. The Phe and Tyr concentrations were calculated from an internal standard ratio. The specific PAH activity was expressed as mU/mg total protein, with mU equal to nmol Tyr produced. The PAH activity of PAH_wild-type COS-7 cells was used as a positive control and set at 100%.
Immunoquantification by Western blot analysis
Small amounts (10–15 μg) of protein lysate from the PAH activity assay were used to detect PAH expression by Western blot. An anti-PAH antibody (Merck Millipore) and an anti-β-actin antibody (Santa Cruz Biotechnology) were used as primary antibodies at a 1:5,000 dilution and 1:2,000 dilution, respectively. A goat anti-mouse IgG-HRP conjugated to horseradish peroxidase (Santa Cruz Biotechnology) was used as the secondary antibody for detection at a 1:10,000 dilution. Signals were generated and recorded using the SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific) as described by the manufacturer’s instructions.
TA cloning and sequencing of the PAH_c.1222C>T variant reversal
Genomic DNA from electroporated PAH_c.1222C>T COS-7 cells was extracted using NucleoSpin Tissue (Macherey-Nagel) according to the manufacturer’s instructions. PCR amplification of the mutated target sites was performed with Q5 High-Fidelity DNA Polymerase (New England Biolabs) at standard conditions. The PCR product was purified with the PureLink PCR Purification Kit (Thermo Fisher Scientific). Poly-A tailing of the purified DNA was completed using Taq Polymerase (Qiagen), followed by cloning into plasmid pMD19-T (Clontech). Upon transformation into E. coli cells and colony growth, DNA was isolated from individual colonies and analyzed by Sanger sequencing, using EF1α forward and IRES reverse sequencing primers.
Sanger sequencing for off-target analysis
The potential off-target sites have been predicted using CasOT (http://eendb.zfgenetics.org/casot/)31, a genome-wide potential Cas9-gRNA off-target searching online tool. The genomic region surrounding the candidate off-target sites were PCR-amplified with Q5 High-Fidelity DNA Polymerase (New England Biolabs) at standard conditions, and the PCR products were purified using PureLink PCR Purification Kit (Thermo Fisher Scientific). Sanger sequencing was utilized to detect their sequences using primers which covered the regions of these sites.
Additional Information
How to cite this article: Pan, Y. et al. CRISPR RNA-guided Fokl nucleases repair a PAH variant in a phenylketonuria model. Sci. Rep. 6, 35794; doi: 10.1038/srep35794 (2016).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Blau, N., Van Spronsen, F. J. & Levy, H. L. Phenylketonuria. Lancet 376, 1417–1427 (2010).
Heintz, C., Cotton, R. G. & Blau, N. Tetrahydrobiopterin, its mode of action on phenylalanine hydroxylase and importance of genotypes for pharmacological therapy of phenylketonuria. Hum Mutat 34, 927–936, 10.1002/humu.22320 (2013).
Blau, N., Shen, N. & Carducci, C. Molecular genetics and diagnosis of phenylketonuria: state of the art. Expert review of molecular diagnostics 14, 655–671, 10.1586/14737159.2014.923760 (2014).
Xie, F. et al. Seamless gene correction of beta-thalassemia mutations in patient-specific iPSCs using CRISPR/Cas9 and piggyBac. Genome Res 24, 1526–1533, 10.1101/gr.173427.114 (2014).
Sander, J. D. & Joung, J. K. CRISPR-Cas systems for editing, regulating and targeting genomes. Nature biotechnology 32, 347–355, 10.1038/nbt.2842 (2014).
Bassuk, A. G., Zheng, A., Li, Y., Tsang, S. H. & Mahajan, V. B. Precision Medicine: Genetic Repair of Retinitis Pigmentosa in Patient-Derived Stem Cells. Sci Rep 6, 19969, 10.1038/srep19969 (2016).
Yin, H. et al. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nature biotechnology 32, 551–553, 10.1038/nbt.2884 (2014).
Nelson, C. E. et al. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 351, 403–407, 10.1126/science.aad5143 (2016).
Long, C. et al. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 351, 400–403, 10.1126/science.aad5725 (2016).
Tabebordbar, M. et al. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 351, 407–411, 10.1126/science.aad5177 (2016).
Fu, Y. et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nature biotechnology 31, 822–826, 10.1038/nbt.2623 (2013).
Kuscu, C., Arslan, S., Singh, R., Thorpe, J. & Adli, M. Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease. Nature biotechnology 32, 677–683, 10.1038/nbt.2916 (2014).
Hara, S. et al. Generation of mutant mice via the CRISPR/Cas9 system using FokI-dCas9. Sci Rep 5, 11221, 10.1038/srep11221 (2015).
Guilinger, J. P., Thompson, D. B. & Liu, D. R. Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nature biotechnology 32, 577–582, 10.1038/nbt.2909 (2014).
Tsai, S. Q. et al. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nature biotechnology 32, 569–576, 10.1038/nbt.2908 (2014).
Pinder, J., Salsman, J. & Dellaire, G. Nuclear domain ‘knock-in’ screen for the evaluation and identification of small molecule enhancers of CRISPR-based genome editing. Nucleic acids research 43, 9379–9392, 10.1093/nar/gkv993 (2015).
Song, J. et al. RS-1 enhances CRISPR/Cas9- and TALEN-mediated knock-in efficiency. Nature communications 7, 10548, 10.1038/ncomms10548 (2016).
Jayathilaka, K. et al. A chemical compound that stimulates the human homologous recombination protein RAD51. Proc Natl Acad Sci USA 105, 15848–15853, 10.1073/pnas.0808046105 (2008).
Strisciuglio, P. & Concolino, D. New Strategies for the Treatment of Phenylketonuria (PKU). Metabolites 4, 1007–1017, 10.3390/metabo4041007 (2014).
van Spronsen, F. J. & Enns, G. M. Future treatment strategies in phenylketonuria. Mol Genet Metab 99 Suppl 1, S90–95, 10.1016/j.ymgme.2009.10.008 (2010).
Hsu, P. D., Lander, E. S. & Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 157, 1262–1278, 10.1016/j.cell.2014.05.010 (2014).
Wang, H., La Russa, M. & Qi, L. S. CRISPR/Cas9 in Genome Editing and Beyond. Annual review of biochemistry 85, 227–264, 10.1146/annurev-biochem-060815-014607 (2016).
Aouida, M. et al. Efficient fdCas9 Synthetic Endonuclease with Improved Specificity for Precise Genome Engineering. PLoS One 10, e0133373, 10.1371/journal.pone.0133373 (2015).
Shen, B. et al. Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nature methods 11, 399–402, 10.1038/nmeth.2857 (2014).
Slaymaker, I. M. et al. Rationally engineered Cas9 nucleases with improved specificity. Science 351, 84–88, 10.1126/science.aad5227 (2016).
Maruyama, T. et al. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nature biotechnology 33, 538–542, 10.1038/nbt.3190 (2015).
Bartholomé, K., Lutz, P. & Bickel, H. Determination of phenylalanine hydroxylase activity in patients with phenylketonuria and hyperphenylalaninemia. Pediatr Res 9, 899 (1975).
Unger, T., Jacobovitch, Y., Dantes, A., Bernheim, R. & Peleg, Y. Applications of the Restriction Free (RF) cloning procedure for molecular manipulations and protein expression. J Struct Biol 172, 34–44, 10.1016/j.jsb.2010.06.016 (2010).
Heigwer, F., Kerr, G. & Boutros, M. E-CRISP: fast CRISPR target site identification. Nature methods 11, 122–123, 10.1038/nmeth.2812 (2014).
Shen, N. et al. Co-expression of phenylalanine hydroxylase variants and effects of interallelic complementation on in vitro enzyme activity and genotype-phenotype correlation. Mol Genet Metab 117, 328–335, 10.1016/j.ymgme.2016.01.004 (2016).
Xiao, A. et al. CasOT: a genome-wide Cas9/gRNA off-target searching tool. Bioinformatics 30, 1180–1182, 10.1093/bioinformatics/btt764 (2014).
Acknowledgements
This work is part of the RD-CONNECT initiative and was supported in part by the FP7-HEALTH-2012-INNOVATION-1 EU Grant No. 305444 (to N.B.) and by two grants from the China Scholarship Council (to N.S. and Y.P.). N.B. and G.F.H. were supported by the Dietmar-Hopp Foundation, St. Leon-Rot. The authors would like to thank Ms. Kathrin Schmidt for technical assistance. The pCMV-FLAG-PAH wild-type plasmid was a generous gift from L. R. Desviat, Madrid, Spain. The FokI-dCas9 plasmid (Addgene plasmid # 52970) was a kind gift from David Liu.
Author information
Authors and Affiliations
Contributions
Y.P. and N.S. conceived and designed the experiments. N.B. coordinated the project. Y.P., N.S., S.J.-K., J.D.H. and C.B. carried out the experiments and analyzed data. Y.P., N.S., J.D.H. and N.B. wrote the manuscript. G.F.H., S.J.-K., C.B. and J.D.H. provided evaluation and manuscript suggestions. Each author contributed to manuscript editing.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Pan, Y., Shen, N., Jung-Klawitter, S. et al. CRISPR RNA-guided FokI nucleases repair a PAH variant in a phenylketonuria model. Sci Rep 6, 35794 (2016). https://doi.org/10.1038/srep35794
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep35794
This article is cited by
-
Genetic etiology and clinical challenges of phenylketonuria
Human Genomics (2022)
-
Deubiquitinase USP19 extends the residual enzymatic activity of phenylalanine hydroxylase variants
Scientific Reports (2022)
-
Modification of improved-genome editing via oviductal nucleic acids delivery (i-GONAD)-mediated knock-in in rats
BMC Biotechnology (2021)
-
Ways of improving precise knock-in by genome-editing technologies
Human Genetics (2019)
-
CRISPR-Cas orthologues and variants: optimizing the repertoire, specificity and delivery of genome engineering tools
Mammalian Genome (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
