
Abstract
We report a biosensing platform for viral load measurement through electrical sensing of viruses on a flexible plastic microchip with printed electrodes. Point-of-care (POC) viral load measurement is of paramount importance with significant impact on a broad range of applications, including infectious disease diagnostics and treatment monitoring specifically in resource-constrained settings. Here, we present a broadly applicable and inexpensive biosensing technology for accurate quantification of bioagents, including viruses in biological samples, such as plasma and artificial saliva, at clinically relevant concentrations. Our microchip fabrication is simple and mass-producible as we print microelectrodes on flexible plastic substrates using conductive inks. We evaluated the microchip technology by detecting and quantifying multiple Human Immunodeficiency Virus (HIV) subtypes (A, B, C, D, E, G and panel), Epstein-Barr Virus (EBV) and Kaposi’s Sarcoma-associated Herpes Virus (KSHV) in a fingerprick volume (50 µL) of PBS, plasma, and artificial saliva samples for a broad range of virus concentrations between 102 copies/mL and 107 copies/mL. We have also evaluated the microchip platform with discarded, de-identified HIV-infected patient samples by comparing our microchip viral load measurement results with reverse transcriptase-quantitative polymerase chain reaction (RT-qPCR) as the gold standard method using Bland-Altman Analysis.
Similar content being viewed by others

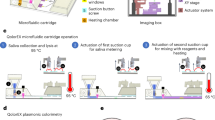
Introduction
The development of sensitive, portable, rapid and cost-effective virus detection and quantification in biological samples has broad diagnostic and biosafety applications in food safety, homeland security and infectious disease diagnosis1,2,3. It has the potential to revolutionize personal healthcare in resource-constrained settings as well as developed countries. The development of such point-of-care (POC) assays for viral load measurement is currently a significant technical and clinical challenge and requires innovative and urgent solutions at the interface of engineering and medicine4,5. Commercially available assays for sensitive viral load measurement, including reverse transcriptase-quantitative polymerase chain reaction (RT-qPCR), require nucleic acid amplification or signal amplification, which are labor-intensive, expensive, time-consuming and complex. Despite the recent advances in micro- and nano-technologies, which have shown a great promise for the development of POC diagnostic platforms, there is currently no commercially available biosensor for POC viral load measurement that is rapid, sensitive, inexpensive and easy-to-use suitable for resource-constrained settings6,7,8,9,10,11,12. A major challenge in developing POC viral load measurement platforms is the sensitive and specific detection and quantification of viruses using a fingerprick volume (<100 µL) of biological samples at clinically relevant concentrations without the need for amplification and sample preprocessing. Here, we present a printed flexible plastic microchip technology that can capture and quantify viruses in a variety of biological samples, including plasma and artificial saliva, through electrical sensing of viral lysate. The integration of electrical sensing with printed flexible plastic microfluidics can potentially provide a promising opportunity to develop an innovative viral load measurement platform that addresses the current clinical challenges associated with viral load testing1,14,15,16,17,18,19,20. To demonstrate the broad applications of this biosensing platform, we evaluated our microchip technology to detect and quantify multiple virus targets, including Human Immunodeficiency Virus-1 (HIV-1) (subtypes A, B, C, D, E, G and panel), Epstein-Barr Virus (EBV), and Kaposi’s Sarcoma-associated Herpes Virus (KSHV) in spiked plasma and artificial saliva samples as well as HIV-infected patient samples.
Results
Viral load measurement – HIV
Figure 1 shows the 3D schematics of the presented technology for bioagent capture and detection on a printed flexible plastic microchip through capacitance spectroscopy of bioagent lysate. To evaluate the ability of the microchip to detect a broad range of targets in biological samples with clinically relevant concentrations, we first demonstrated HIV viral load measurement in a fingerprick volume (50 µL) of spiked-Phosphate Buffered Saline (PBS) and plasma samples for multiple HIV-1 subtypes (A, B, C, D, E, G and panel) ( Fig. 2 ). HIV-1 particles were captured by biotinylated polyclonal anti-gp120 antibodies anchored to streptavidin-coated magnetic beads ( Fig. 1 ). Captured viruses were washed with a low-electrically conductive solution (10% glycerol) and lysed using 1% Triton x-100. The virus lysis step releases the charged molecules into the lysis buffer, i.e. Triton x-100, which changes the bulk electrical properties of the solution. Captured viruses were detected through capacitance spectroscopy of viral lysate samples on a flexible plastic microchip with screen-printed electrodes. Our preliminary experimental results, using diluted PBS in deionized (DI) water samples, demonstrated that the impedance magnitude of the samples decreases, while the capacitance magnitude increases due to an increase in electrical conductivity of the samples (Supplementary Fig. S1a, b). These results also demonstrated that capacitance spectroscopy in our microchips provided a more sensitive detection method as compared to impedance spectroscopy at 1,000 Hz (Supplementary Fig. S1c). Moreover, these results show that the maximum capacitance magnitude change in the samples occurred at frequencies between 100 Hz and 1,000 Hz ( Supplementary Fig. S2 ). We chose 1,000 Hz as the detection frequency for viral load measurements as we observed lower noise at this frequency. We measured the capacitance magnitude response due to virus lysis at 2V and 1,000 Hz in HIV-spiked PBS ( Supplementary Fig. S3 ) and HIV-spiked plasma ( Fig. 2 ) samples with clinically relevant virus dilutions between 102 copies/mL and 107 copies/mL. The lowest diluted virus concentrations in HIV-spiked PBS samples with statistically significant capacitance change compared to the control samples were 103 copies/mL (p = 0.0005, n = 8), 102 copies/mL (p = 0.0104, n = 8), 103 copies/mL (p = 0.0007, n = 8), 103 copies/mL (p = 0.0002, n = 8), 102 copies/mL (p = 0.007, n = 8), 103 copies/mL (p = 0.002, n = 8), and 103 copies/mL (p = 0.010, n = 8) for subtypes A, B, C, D, E, G and panel, respectively (Fig. S3).

3D Schematic of pathogen capture and detection on a printed flexible plastic microchip.
(a) Magnetic beads conjugated with antibodies to capture HIV, EBV, KSHV or E. coli are mixed with a fingerprick volume of a biological sample off-chip. (b) Captured pathogens are washed 4 times with a low conducting media (glycerol) to remove residual high electrically conductive background. (c) The captured bioagents are then lysed with Triton x-100 (d) The pathogen lysate is then injected into the chip for capacitance measurement. The captured and isolated pathogens are detected through capacitance spectroscopy of the lysate on a printed flexible plastic microchip.

Microchip evaluation using HIV-1 subtypes spiked in plasma.
Linear correlation between viral load of the samples and on-chip capacitance measurements at 2 V and 1 KHz for multiple HIV-1 subtypes A (a), B (b), C (c), D (d), E (e), G (f) and panel (g) showed coefficient of determinations between 0.83 and 0.94. The lowest dilutions that showed significant capacitance change compared to virus-free control plasma samples were 103 (p = 0.001, n = 8), 103 (p = 0.008, n = 8), 102 (p = 0.049, n = 8), 102 (p = 0.0002, n = 8), 103 (p = 0.002, n = 8), 103 (p = 0.0007, n = 8) and 104 (p = 0.004, n = 8) for viral load measurement in plasma samples spiked with HIV-1 subtypes A (a), B (b), C (c), D (d), E (e), G (f), and panel (g), respectively. The limits of detection were established as the lowest concentration from the experimental results with a statistically significant difference (p<0.05) from virus-free control sample calculated through Mann-Whitney method of analysis. (h) The general trend of all subtypes of HIV-spiked plasma samples was obtained by generating a simple linear regression using all of the experimental results obtained from HIV-1 subtypes A, B, C, D, E, G and panel. Error bars represent standard error of mean. Control samples were pathogen-free plasma samples. Horizontal black and red dotted lines represent the average capacitance magnitude of the control samples and control ± standard error, respectively.
We also observed that the lowest diluted virus concentrations in HIV-spiked plasma samples with statistically significant capacitance change compared to the control samples were 103 copies/mL (p = 0.001, n = 8), 103 copies/mL (p = 0.008, n = 8), 102 copies/mL (p = 0.049, n = 8), 102 copies/mL (p = 0.0002, n = 8), 103 copies/mL (p = 0.002, n = 8), 103 copies/mL (p = 0.0007, n = 8), 104 copies/mL (p = 0.004, n = 8) for HIV-1 subtypes A, B, C, D, E, G and panel, respectively ( Fig. 2 ). The different detection limits for HIV subtypes may be due to the differences between the HIV-1 gp120 envelope sequences of the subtypes used in this study as compared to the sequence of the immobilized anti-gp120 antibody used for virus capture. To demonstrate this difference, we report the gp120 sequence of the HIV subtypes A, B, D, E and G aligned with the EU541617 reference strain used to generate the polyclonal anti-gp120 antibody (Supplementary Fig. S4). The inherent cumulative inaccuracy of serial dilution can affect the coefficients of determination of our samples making limit of detection determination challenging.
Viral load measurement - EBV and KSHV
To demonstrate the broad applicability of the microchip technology, we also evaluated the platform to detect and quantify EBV and KSHV spiked in PBS and artificial saliva samples. EBV and KSHV particles were selectively captured on streptavidin-coated magnetic beads conjugated with biotinylated MA-gp350/250 and MAb-K ORF 8.1A/B antibodies, respectively. The linear correlation between the capacitance magnitude of the viral lysate and viral load of PBS and artificial saliva samples spiked with EBV and KSHV are reported in Figure 3 . The samples were spiked with viruses at concentrations ranging from 102 copies/mL to 107 copies/mL. The lowest diluted virus concentrations in EBV-spiked PBS and artificial saliva samples with statistically significant capacitance change compared to the control samples were 104 copies/mL (p = 0.0070, n = 8) and 102 copies/mL (p = 0.0007, n = 8), respectively ( Fig. 3a, b ). We also observed that the lowest diluted virus concentrations in KSHV-spiked PBS and artificial saliva samples with statistically significant capacitance change compared to the control samples were 102 copies/mL (p = 0.0426, n = 8) and 103 copies/mL (p = 0.0047, n = 8), respectively ( Fig. 3c, d ). Therefore, the presented microchip technology satisfies the clinical requirement to detect EBV and KSHV21,22.

Microchip evaluation using EBV and KSHV spiked in PBS and saliva and E.coli spiked in saliva samples.
Linear correlations between viral load of samples and on-chip capacitance measurements at 2 V and 1 KHz for EBV-spiked PBS (a) and saliva (b) samples were calculated. The lowest dilutions with significant capacitance change as compared to virus-free control samples were 104 copies/mL in EBV-spiked PBS (a, p = 0.0070, n = 8) and 102 copies/mL in EBV-spiked saliva (b, p = 0.0007, n = 8) samples. Linear correlations between viral load of samples and on-chip capacitance measurements at 2 V and 1 KHz for KSHV-spiked PBS (c) and saliva (d) samples were calculated. The lowest dilutions with significant capacitance change as compared to virus-free control samples were 102 copies/mL (p = 0.0426, n = 8) and 103 copies/mL (p = 0.0047, n = 8) for KSHV-spiked PBS and saliva samples, respectively. Error bars represent standard error of mean. (e)E. coli was spiked in artificial saliva. 5% Triton was used in contrast to the 1% Triton used for viral lysis. The lowest statistically significant dilution of stock is 104 (p = 0.0013, n = 8) and was calculated utilizing Mann-Whitney analysis. Error bars represent SEM. Control samples were pathogen-free (a, c) PBS and (b, d, e) artificial saliva samples. Horizontal black and red dotted lines represent the average capacitance magnitude of the control samples and control ± standard error, respectively.
Detection of bacterial pathogens
This microchip technology has the potential to selectively detect pathogens other than viruses, including bacterial pathogens. Here, we present our results on capture and detection of E. coli in artificial saliva using the printed flexible plastic microchips. E. coli cells were selectively captured on magnetic beads conjugated with anti-E. coli LPS antibody in 50 µL of diluted artificial saliva samples. The lowest diluted cell concentration in E. coli-spiked artificial saliva samples with statistically significant capacitance change compared to the control samples was 104 cells/mL (p = 0.0013, n = 8) ( Fig. 3e ).
Specificity evaluation
To evaluate the specificity of the presented microchip technology, we tested the platform to detect a target virus such as HIV or KSHV in the presence of other bioagents such as EBV and E. coli. We have previously shown that HIV can be specifically detected in the presence of other viruses, including EBV in samples using electrical sensing of viral lysate on-chip9. Here, we also studied if the presence of non-target pathogens such as E. coli, which are much larger in size, may release a significantly higher amount of ions into the solution. Control samples in our specificity evaluation experiments were pathogen-free plasma and artificial saliva samples. Figure 4a shows the specificity evaluation results for HIV detection in plasma samples in the presence of E. coli. In this experiment, we used magnetic beads conjugated with anti-gp120 antibody to selectively capture HIV. The capacitance magnitude of E. coli samples without HIV was not statistically different than control samples (p>0.05, n = 4) with a statistical power of 0.68. However, the capacitance magnitude of the mixture of HIV subtype B and E. coli was significantly different than control and E. coli samples (p<0.05, n = 4) ( Fig. 4a ). These results showed that anti-gp120 antibody specifically captured and isolated HIV and not E. coli in the samples. Figure 4b shows the specificity evaluation of the microchip for KSHV detection in the presence of EBV and E. coli. We used anti-ORF K 8.1 A/B antibody for microchip specificity evaluation to detect KSHV. The capacitance magnitude of the mixture of E. coli and EBV did not show a statistically significant difference (p>0.05, n = 4) compared to the control samples with a statistical power of 0.73, whereas the capacitance magnitude of the mixture of KSHV, EBV, and E. coli was significantly different than control samples, as well as the mixture of EBV and E. coli samples (p<0.05, n = 4) ( Fig. 4b ). This is due to highly specific capture and isolation of KSHV in the samples using anti-ORF K 8.1 A/B antibody.

Specificity evaluations of the microchip.
(a) HIV-1 subtype B was specifically captured and detected in the presence of E. coli spiked in plasma samples. E. coli at concentrations of 104 cells/mL and 106 cells/mL showed no significant difference (p>0.05, n = 4) against HIV-free control. HIV-spiked samples (104 copies/mL and 105 copies/mL) and the mixture of HIV-1 and E. coli samples showed statistically different (p<0.05, n = 4) capacitance values compared to virus-free control samples. (b) KSHV was specifically captured and isolated from a mixture of KSHV, EBV and E. coli in artificial saliva. Mixtures of EBV (103 copies/mL and 105 copies/mL) and E. coli (103 cells/mL and 105 cells/mL) showed no significant difference (p>0.05, n = 4) in capacitance magnitude from KSHV-free control samples. KSHV-spiked samples at concentrations of 103 copies/mL and 105 copies/mL showed a significant difference (p<0.05, n = 4) from control. The capacitance magnitude of the mixture of KSHV, EBV and E. coli samples were also significantly different than control samples. Brackets indicate statistically significant capacitance magnitude shift between the linked groups. Control samples were pathogen-free (a) plasma and (b) artificial saliva samples. Error bars represent standard error of mean (n = 4).
Patient samples
We also evaluated our viral load measurement using five anonymous, discarded HIV-infected patient plasma samples. We first generated a standard curve by performing a least-square fit linear regression for HIV viral load measurement using the experimental results with the HIV-spiked samples reported in Supplementary Figures S3a-g ( Fig. S3h ). We used this standard curve to translate the capacitance magnitude measurement to viral load of the patient samples tested in this study. The quantitative HIV viral load measurement results for the five patient samples using our microchip platform and RT-qPCR are presented in Supplementary Table S2 . The capacitance magnitude change for one of the patient samples, which had undetectable viral load by means of RT-qPCR, was statistically negligible as compared to the control samples in our microchip. The data point related to the viral load of this patient sample is shown at the origin of Figure 5a . These results were analyzed and statistically evaluated using the Bland-Altman method23,24 and presented in Figure 5b . These results confirmed that there was no evidence for a systematic bias for viral load measurement in HIV-infected patient samples using our printed flexible plastic microchip technology.

Device validation with patient samples.
(a) Correlation data of microchip viral load and the viral load obtained through RT-qPCR. Viral load of zero was considered for patients with undetectable loads. Error bars represent standard error of mean (n = 8). (b) Bland-Altman comparison of viral loads obtained through RT-qPCR and our printed flexible plastic microchip. Bland-Altman analysis did not show any evidence of systematic bias. Quantitative information is available in supplementary Table S2.
Discussion
We have previously shown that impedance magnitude of viral lysate can be used for virus detection on microfluidic devices with gold microelectrodes fabricated on glass substrates using lift-off lithography method9. We detected HIV-1 spiked in PBS samples with virus concentrations on the order of 108 copies/mL. Here, we demonstrate a new microchip technology for virus detection and quantification by integrating screen-printing silver microelectrodes on flexible plastic substrates and capacitance spectroscopy. Double layer capacitance at low frequencies, such as 1,000 Hz, in electrically low conductive solutions, such as the viral lysate samples used in this work, changes more significantly as the ion concentration in the sample increases compared to impedance magnitude25. We have evaluated our technology using multiple bioagents, including HIV, EBV, KSHV and E. coli. Our results show that this microchip technology can detect viruses in complex biological samples, such as human plasma and artificial saliva, with clinically relevant virus concentrations. This microchip technology can be used for acute HIV detection, when maximum viral replication occurs26, as well as for treatment failure diagnosis based on POC ART monitoring defined by the World Health Organziation (WHO)13. Further technology development is required to enhance the sensitivity comparable with PCR-based assays. This biosensing platform can be potentially used for detection and quantification of other viral particles that have well-described biomarkers and antibodies for capture, including herpes, influenza, malaria and tuberculosis.
The presented microchip technology is a platform technology to detect and quantify multiple viruses such as HIV, EBV and KSHV. It can potentially detect multiple viruses at the same time and on a single chip with parallel microchannels. In such a device, each microchannel has a separate pair of microelectrodes for electrical measurement and is functionalized with a specific antibody to capture a target virus for detection. Such microchip design enables multiplexing, however, the cost per test increases accordingly. Co-infections with other pathogens such as EBV, Cytomegalovirus (CMV), Human Papillomavirus (HPV), Herpes simplex virus (HSV), and Candida are also common in HIV-infected patients, specifically in those not receiving ART. Therefore, multiplexing on a single chip to detect these pathogens at the same time can have a significant impact on HIV management in developing countries. Future studies can evaluate the specificity of the electrical sensing microchip technology to detect HIV in the presence of CMV, HPV, HSV and Candida in plasma.
To prepare this microchip technology for field-testing, further technology development and optimization is required to perform all of the sample handling steps, including virus capture, washing and lysis on-chip. It is also important to evaluate the effect of temperature, humidity and stability of reactions at different sites on the performance of this microchip technology with respect to sensitivity, specificity and reproducibility. Device throughput for viral load testing is also another design parameter that should be addressed during technology transfer and commercialization.
The electrical measurements in this study were performed using an impedance spectrometer to evaluate the performance of the microchip technology for AC frequencies between 100 Hz and 1 MHz. These results show that the electrical measurements can be done at a single frequency for multiple viruses, i.e. 1,000 Hz. Therefore, it is potentially possible to build a hand-held impedance/capacitance meter utilizing affordable miniaturized frequency oscillators, microcontrollers and impedance/capacitance meter microchips available in the market.
Here, we evaluated our printed flexible plastic microchip with respect to sensitivity, specificity and linearity. To validate this technology, other parameters, including repeatability, robustness, precision, system suitability, and performance over time should also be evaluated. Furthermore, it is also required to evaluate the technology with other HIV subtypes, including the less common members of group M (K, J, F, H, I), N and O, as well as HIV-2 (groups A and B).
Viral load measurement is critical in infectious disease diagnosis and treatment monitoring and can facilitate treatment expansion especially in resource-constrained settings. ART is a successful treatment to suppress HIV, prolong life in HIV-infected patients and reduce HIV infection transmission rates, and has become even more affordable and accessible in developing countries27. However, a majority of HIV-infected individuals do not receive ART due to lack of affordable, rapid and sensitive HIV diagnostic tools at the POC28. Virological suppression represents the most accurate immunological response of the patients to ART29. Thus, the most accurate and preferred method for ART monitoring to diagnose treatment failure, as recommended by the 2013 consolidated guidelines on the use of ART, is viral load measurement30. Viral load testing is important for identifying treatment failure and enabling medication changes or other interventions to prevent immunologic compromise and clinical progression of disease. POC viral load tests could be used to determine the efficacy of second line antiretroviral therapies in addition to identifying failure of initial regimens. Early detection of virological failure allows for both targeted adherence interventions and better preservation of the efficacy of second-line regimens. In addition, POC viral load testing may also be applied to the diagnosis of acute HIV infection and prompt initiation of ART. Prompt ART initiation may lead to a greater preservation of immune function and has the potential to reduce HIV-1 transmission. Currently available RNA- and PCR-based assays for viral load measurement in developed countries are expensive, laboratory-based, complex, time-consuming, and labor-intensive and cannot be easily used in resource-constrained settings. p24 assays and CD4+ cell counting strategy cannot be used for monitoring the efficacy of treatment and detecting ART failure in patients31,32,33,34,35. Therefore, to detect early virological failure and to expand access to ART, new emerging technologies are urgently needed to facilitate viral load testing at the POC36,37,38,39,40. POC viral load testing may lead to increasing the adherence and minimizing the drug resistance through timely detection of virological failure41,42,43.
Kaposi’s Sarcoma-associated Herpes Virus (KSHV) and Epstein-Barr Virus (EBV) are human gamma herpesviruses44. KSHV and EBV are the causative agents of a number of malignancies, especially common in immunosuppressed individuals such as those with HIV/AIDS. KSHV is the etiologic agent of Kaposi’s sarcoma (KS)45,46,47 and primary effusion lymphoma (PEL)48,49 and is strongly associated with multicentric Castleman’s disease50,51, an aggressive lymphoproliferative disorder. KS is epidemic in sub Saharan Africa and is the leading AIDS malignancy52,53,54. EBV has a causative role in several human malignancies, including Burkitt's lymphoma55, Hodgkin's lymphoma56, nasopharyngeal carcinoma57, lymphoproliferative disease in immune-suppressed patients58 and lymphomas in HIV-infected patients59. There are no specific therapies available for these cancers and therapy generally relies on cytotoxic chemotherapy or improvement of immune function. KSHV is frequently found in saliva of infected persons and oral contact is the most common route of transmission in endemic regions60,61,62. EBV also spreads through saliva and may initially replicate in the oropharyngeal epithelium59. However, the details of transmission are poorly understood. Rapid and efficient detection of KSHV and EBV in saliva would enable studies to better understand viral transmission. Such an understanding may lead to strategies to interrupt transmission of these viruses and prevent virus-associated malignancies. Detection of viral load in blood can also be useful to anticipate the onset of KSHV- and EBV-associated diseases or to monitor the response to therapies. In fact, elevated KSHV viral load in blood has been shown to predict the progression of KS63,64. In addition, several studies have shown a strong correlation between EBV burden in blood and presence of post-transplant lymphoproliferative disease (PTLD) in transplantation recipients65,66,67. Elevated EBV DNA in patients can be detected prior to PTLD diagnosis68.
Electrical sensing in microfluidics is insensitive to light intensity and does not need the bulky equipment usually required in optical sensing platforms and is a powerful modality to create portable and sensitive biosensors2,3,9,69,70. Paper-based microfluidics has shown great promise in developing affordable, mass-producible and disposable POC diagnostic assays for developing countries14,71. Paper substrates are light (~10 mg/cm2), thin, flexible and disposable, and paper microchips can be fabricated for a few cents14,72,73,74. Here, we fabricated our microchips by screen-printing conductive silver ink on flexible hydrophobic plastic sheets that are thin, light, flexible, and inexpensive. These advantages are similar to those of paper-based microfluidics. In addition, our chip fabrication is less complicated as we do not use commonly used methods for paper-based microfluidic fabrication, including photolithography19, plotting75, inkjet etching76, plasma etching77, or wax printing78,79. We report a microchip technology that integrates electrical sensing and printed flexible plastic microfluidics for inexpensive and rapid viral load measurement through viral lysate capacitance spectroscopy. The material cost of the microchip include less than 1 cent for flexible plastic sheets, less than 10 cents for electrodes and less than 1 cent for DSA. The material costs for magnetic beads and antibody (anti-gp120 antibody) are 80 cents and 87 cents per test, respectively. These costs do not include labor costs. Therefore, we present a new viral load testing technology with the material cost per microchip of less than $2. The current assay time in the laboratory can take up to an hour, including 30 minutes for sample incubation to capture viruses, 4 minutes for washing, 1 minute for virus lysis, 10 minutes for read-out and analysis and transfer steps. In this microchip technology, viruses are captured and isolated from biological samples off-chip utilizing highly specific antibodies. The virus lysis step in this method releases charged molecules in viral particles into an electrically low conductive solution, i.e. Triton x-100. These charged molecules change the electrical properties of the bulk solution as well as double layer properties on the surface of the electrodes. Capacitance spectroscopy of the viral lysate solution on-chip reveals the virus concentration in the sample.
There are a variety of inhibitory factors with different concentrations in biological samples such as plasma, saliva and semen. These inhibitory factors may have interference with the infectivity of the virus in body fluids. For instance, HIV can be reproduced and cultured in tissues and most of the body fluids such as plasma, breast milk and genital secretions, except saliva80. Saliva is able to specifically suppress HIV-180. However, saliva does not have a significant effect on the infectivity of other viruses such as EBV, Herpes simplex virus (HSV) and cytomegalovirus (CMV)81,82,83. Saliva is also the vehicle of person-to-person KSHV spread60,84,85. Inhibitory factors such as highly charged sulfated polysaccharides, secretory leukocyte protease inhibitor (SLPI), fibronectin and thrombospondin 1 (TSP1) in whole saliva may interfere with the infectivity of HIV-1 through altering the CD4-gp120 interactions80,86. Such inhibitory factors may be also present in plasma, but in very low concentrations80,87. KSHV levels in blood can be quite variable, depending on the level of a patient's immunosuppression and burden of disease88. Furthermore, inhibitory factors may interfere in enzyme-based assays or nucleic acid-based amplification methods. In our electrical sensing technology, we first capture and isolate intact viruses from artificial saliva or plasma using antibodies. The captured viruses are then washed several times to remove non-target bioagents, including inhibitory factors, before viral lysis and detection. The electrical sensing assay does not rely on enzyme-based processing or amplification of nucleic acids. We have not observed interference from such inhibitory factors in our studies using HIV-spiked plasma samples as well as HIV-infected patient plasma samples. However, further evaluation is required to investigate the effect of inhibitory factors on the performance of the electrical sensing microchip technology. Viral load level in different body fluids such as plasma and saliva is dynamic and may or may not be correlated with each other89. For example, HIV load level in plasma and semen are correlated and can be influenced by multiple factors such as antiretroviral therapy and adherence, HIV treatment resistance and the stage of HIV disease89. Also, HIV loads in plasma and saliva are poorly correlated90.
Materials and Methods
Reagents
Ultrapure grade (Type I) water was obtained through purification using Milli-Q® Academic A-10® (Millipore). Phosphate buffered saline (PBS) was obtained from Gibco (Grand Island, NY; 10010). Polyclonal biotinylated goat anti-gp120 (4.0 mg/mL) for HIV-1 capture was purchased from Abcam (Cambridge, MA; ab53937). Mouse monoclonal antibodies reactive to proteins encoded by ORF K 8.1 A/B (0.86 mg/mL) of HHV-8 was purchased from Advanced biotechnologies (Columbia, MD; 13-212-100). Mouse monoclonal antibodies reactive to 350/220 KDa viral glycoprotein of EBV were purchased from Millipore (Billerica, MA; MAB10219). Mouse monoclonal antibodies reactive to E. coli lipopolysaccharide (0.5 mg/mL) for E. coli capture were acquired from Abcam (Cambridge, MA; AB35654). Glycerol (56-81-5) and Triton x-100 (9002-93-1) were both purchased from Sigma (St. Louis, MO). RPMI-1640 with L-Glutamine (10-040) was purchased from Corning (Manassas, VA). Fetal bovine serum (100-106) was obtained from Gemini, penicillin/streptomycin (15070-063) was purchased from Invitrogen, HEPES buffer (15630-080) was purchased from Life Technologies (Grand Island, NY), and recombinant human interleukin-2 (11011456001) was purchased from Roche.
Preparation of R20/IL-2
R20/IL-2 contains RPMI-1640 with L-Glutamine (300 mg/mL), penicillin/streptomycin (50 U/mL 50 μg/mL), fetal bovine serum (heat inactivated) 20%, 10 mM of HEPES buffer, and 100 U/mL of recombinant human interleukin-2.
Artificial saliva preparation
Artificial saliva prepared for these experiments has the following composition: 0.1 L each of 25 mM K2HPO4 (Sigma, 7758-11-4), 24 mM Na2HPO4 (Sigma, 7558-79-4), 150 mM KHCO3 (Sigma, 298-14-6), 1.5 mM MgCl2 (Sigma, 7786-30-3), 0.1 L of 100 mM NaCl (Sigma, 7647-14-5), 0.006 L of 25 mM citric acid (Sigma, 77-92-9) and 0.1 L of 5 mM CaCl2 (Sigma, 10043-52-4). The pH of the solution was maintained at 6.7 through the addition of NaOH (Sigma-Aldrich, 1310-73-2) or HCl (Sigma, 7647-01-0). Although this artificial saliva solution does not have proteins and enzymes, which are present in saliva making it a complex fluid, the artificial saliva prepared here is a model system that mimics ionic strength and ionic contents in saliva.
HIV-spiked plasma samples preparation
Human whole blood from HIV-free donors was acquired from Research Blood Components, LLC, Cambridge, MA. Plasma was separated from whole blood samples through centrifugation at 1,500 rpm for 15 minutes. Plasma samples were spiked with viruses at stock concentration of 108 copies/mL and serially diluted to prepare samples with diluted virus concentrations between 102 copies/mL and 107 copies/mL.
HIV-infected patient samples
Discarded, de-identified clinical research plasma specimens from HIV-infected patients were used in this study. Informed consent was obtained from all patients prior to sample collection. Blood was collected by peripheral venipuncture. After initial centrifugation at 1,500 rpm for 15 minutes to separate the cellular and plasma components, the plasma was removed and centrifuged an additional time to remove possible debris. The resulting plasma was frozen and stored at −80°C until use. Patient samples (1 mL) were sent to the Infectious Diseases Clinic at the Brigham and Women’s Hospital for viral load measurement using Roche-COBAS AmpliPrep/COBAS TaqMan HIV-1 test, v2.0. The study protocol involving human subjects was approved by the Institutional Review Board of the Brigham and Women’s Hospital, Harvard Medical School. All experiments involving human subjects were performed in accordance with the relevant approved guidelines and regulations.
Printed flexible plastic microchip fabrication
Transparency sheets (3M, St. Paul, Minnesota, CG5000) were used as the substrates of the microchip. With the use of a laser cutter (Universal Laser systems Inc., VLS 2.3, Scottsdale, AZ), inlet and outlets were cut into transparency sheets with a radius of 0.65 mm91. The power and scan rate at a height of 3.175 mm were at 83 W and 6 mm/s, respectively. The channel (12 mm × 5 mm) was cut into a double-sided adhesive (DSA; 3M; 8213) of 80 µm thickness. The power and scan speed were 10 W and 6 mm/s respectively, with the laser at a height of 3.175 mm. Silicone adhesive (Loctite, Rocky Hill, CT; 341-882) and silver-vinyl ink (<0.015 Ω/sq/mil; ECM, Delaware, OH; CI-1001) pastes were mixed in a ratio of 1:5 (w/w) to make silver paste. The dimensions of the electrodes (2 cm × 2 mm and separated by 2 mm) were cut into the masking paper (Mask-ease, Blick Art Materials; 44908-1003) with the laser cutter. The masking paper was taped over the transparency sheet and the silver paste mixture was applied and was smeared using a glass slide to create a uniform layer of the ink on top of the paper substrate (Supplementary Fig. S5). The masking tape was removed and the sheet was baked in an oven at 175°C for 3 hours. The transparency sheet with the printed electrodes was used as a base substrate of the microchip. The DSA layer, with the microchannel designs cut on it, was then attached to the paper substrate with printed electrodes and another transparency paper was attached to the other side.
Magnetic bead preparation
200 µL of streptavidin-coated magnetic beads (Thermo Scientific, Rockford, IL; 88817) of 1 µm diameter was washed three times with PBS in a microcentrifuge tube. A Magna GrIP™ (Millipore) magnetic stand was used to isolate magnetic beads during washing. 10 µL of biotinylated target antibody was added to the solution. The mixture was incubated over-night on a shaker at 4°C.
Target bio-agent capture with conjugated beads
The antibody conjugated magnetic beads were washed twice with PBS and resuspended in 2 mL PBS to remove any unconjugated antibody in the solution. 50 µL of magnetic beads was taken in a microcentrifuge tube. The supernatant was removed with the help of Magna GrIP™ and was replaced with 50 µL of the pathogen-spiked PBS, plasma and plasma sample. This sample-bead mixture was incubated at room temperature for half an hour on a rotator (15 rpm). Control samples for the experiments were prepared by mixing 50 µL of virus-free PBS/saliva/plasma with magnetic beads.
Washing and viral lysis
The sample-bead mixture was washed with 10% (v/v) glycerol 4 times off-chip following the room temperature incubation. The washing step effectively removes electrically conductive solution from the sample. This is critical in reducing sources of noise in the capacitance measurement results. The captured viruses were then lysed off-chip by replacing the glycerol with 50 µL of 1% (v/v) Triton X-100, which helps in breaking down the lipid bilayer. The sample was incubated at room temperature with 1% Triton X-100 for 1 minute prior to capacitance measurement on-chip.
Capacitance measurement
20 µL of viral lysate was injected into the printed flexible plastic microchip and capacitance was measured using an LCR meter (LCR8110G, GWInstek, CA) for frequencies between 100 Hz and 1 MHz and 2 V.
HIV culture
NIH provided all HIV-1 subtypes (A, B, C, D, E and G) and panel (circulating recombinant forms, CRF01_AE and CRF02_AG) under the research and reagent program. All subtypes were isolated from both Uganda and the United States. The panel of isolates contains six strains of HIV-1 that are globally prevalent. Standard co-culture methods were used to prepare viral stocks. Peripheral blood mononuclear cells (PBMC) were extracted from donors (HIV-1 negative) using the Ficoll Hypaque technique. PBMCs extracted from both HIV negative and HIV positive donors were co-cultured after stimulation with PHA (0.25 µg/mL) for 3 days. 100 U/mL of R20/IL-2 was used to maintain cultures incubated at 37°C with 5% CO2 atmosphere. Culture supernatants were replaced by fresh medium once every week. p24 levels of the supernatant were tested and when they reached 20 ng/mL, cultures were terminated. Viral stocks were serially diluted in PBS and plasma to prepare spiked samples with virus concentrations ranging from 102 copies/mL to 107 copies/mL. Roche-COBAS AmpliPrep/COBAS TaqMan HIV-1 test, v2.0 was used for viral load measurement in the stock virus samples at the Brigham and Women’s Hospital.
KSHV and EBV production
iSLK.21992 cells (Gift from Don Ganem) were cultured in DMEM medium supplemented with 10% bovine growth serum (BGS) (HyClone) and 15 μg/ml gentamicin. iSLK.219 cells were cultured in the presence of 1 μg/mL puromycin and 250 μg/ml G418. B958 cells were cultured in RPMI medium supplemented with 10% bovine growth serum (BGS) (HyClone) and 15 μg/mL gentamicin.
Recombinant KSHV.BAC219 virus stocks were prepared from stable iSLK-219 cells induced by doxycycline (1 μg/mL) in the absence of puromycin and G418. Three days later, supernatant was collected and cleared of cells and debris by filtration (0.45 μm). Virus particles were pelleted by centrifugation (13,300 g for 2 h at 4°C) using a Sorvall SLA-600TC rotor.
Infectious virus was assessed by titration of supernatants using infection of 293T cells. Cell-free virus supernatant was diluted in fresh medium (2 mL final per well) and used to inoculate 293T cells seeded at approximately 3.5 × 105 cells/well in 6-well plates 24 h prior infection. Following inoculation, plates were immediately centrifuged (600 × g for 1h at 25°C) and supernatant was removed and replaced with fresh medium. The percentage of GFP-positive cells (from GFP-KSHV) was analyzed 48h later by fluorescence-activated cell sorting (FACS). Infectious units (IU) are expressed as the number of GFP-positive cells per mL of cell-free virus supernatant 48h after infection.
EBV virus stocks were prepared from B95893 induced by TPA (20 ng/mL) and sodium butyrate (32 mM). One day after induction, cells were centrifuged and media was replaced by fresh RPMI. Six days later, supernatant was collected and cleared of cells and debris by filtration (0.45 μm). Virus particles were collected by centrifugation (13,300 g for 2 h at 4°C) using a Sorvall SLA-600TC rotor.
E. coli sample preparation and lysis conditions
E. coli was cultivated from the DH5α strain by incubating them overnight at 37°C in Luria-Bertani (LB) medium that is composed of 10 g/L of tryptone, 5 g/L of yeast extract and 10 g/L of NaCl. E.coli was separated by centrifugation at 10,000 g for 3 minutes and suspended in freshly prepared artificial saliva. E.coli lysis was performed with the same viral lysis protocol (see methods section) but with 5% Triton x-100 replacing 1% Triton x-100 as the lysing agent. Incubation after the addition of 5% Triton x-100 was at 60°C to improve the lysis of E.coli.
Statistical analysis
The normality of the data collected for capacitance measurement was analyzed using Anderson-Darling test. Since the data is not normal, we selected non-parametric test for statistical analysis. Viral load comparisons between groups were analyzed using nonparametric Mann Whitney t test with threshold set at 0.05 (p< 0.05). Error bars represent standard error of the mean (SEM). We observed statistically significant difference with the sample sizes used in this study to evaluate the limit of detection (n = 4) of the microchip as well as microchip specificity (n = 8). A comparison study was performed as well with Bland-Altman analysis, between results from the printed flexible plastic microchips and results from RT-qPCR. Statistical analysis was performed using Prism (Version 5, Graphpad, La Jolla, CA).
References
Yetisen, A. K., Akram, M. S. & Lowe, C. R. Paper-based microfluidic point-of-care diagnostic devices. Lab. Chip. 13, 2210–2251 (2013).
Shafiee, H., Caldwell, J. L., Sano, M. B. & Davalos, R. V. Contactless dielectrophoresis: a new technique for cell manipulation. Biomed. Microdevices 11, 997–1006 (2009).
Shafiee, H., Sano, M. B., Henslee, E. A., Caldwell, J. L. & Davalos, R. V. Selective isolation of live/dead cells using contactless dielectrophoresis (cDEP). Lab. Chip. 10, 438–445 (2010).
Shafiee, H. et al. Emerging technologies for point-of-care management of HIV infection. Annu. Rev. Med. 66, 14.1–14.19 (2014).
Wang, S., Xu, F. & Demirci, U. Advances in developing HIV-1 viral load assays for resource-limited settings. Biotechnol Adv. 28, 770–781 (2010).
Wang, S. et al. Efficient on-chip isolation of HIV subtypes. Lab. Chip. 12, 1508–1515 (2012).
Tokel, O., Inci, F. & Demirci, U. Advances in plasmonic technologies for point of care applications. Chem. Rev. 114, 5728–5752 (2014).
Shafiee, H. et al. Nanostructured optical photonic crystal biosensor for HIV viral load measurement. Sci. Rep. 4, e4116 (2014).
Shafiee, H. et al. Acute on-chip HIV detection through label-free electrical sensing of viral nano-lysate. Small 9, 2553–2563 (2013).
Inci, F. et al. Nanoplasmonic quantitative detection of intact viruses from unprocessed whole blood. ACS. Nano 7, 4733–4745 (2013).
Whitesides, G. M. Cool, or simple and cheap? Why not both? Lab. Chip. 13, 11–13 (2013).
Blow, N. Microfluidics: the great divide. Nat. Methods 6, 683–686 (2009).
World Health Organization, The use of antiretroviral drugs for treatment and preventing HIV infection, (2013). . Available at: http://apps.who.int/iris/bitstream/10665/85321/1/9789241505727_eng.pdf?ua = 1. Date of access: 28/08/2014.
Martinez, A. W., Phillips, S. T., Whitesides, G. M. & Carrilho, E. Diagnostics for the developing world: microfluidic paper-based analytical devices. Anal. Chem. 82, 3–10 (2010).
Schilling, K. M., Lepore, A. L., Kurian, J. A. & Martinez, A. W. Fully enclosed microfluidic paper-based analytical devices. Anal. Chem. 84, 1579–1585 (2012).
Martinez, A. W. Microfluidic paper-based analytical devices: from POCKET to paper-based ELISA. Bioanalysis 3, 2589–2592 (2011).
Cheng, C. M. et al. Paper-based ELISA. Angew. Chem. Int. Ed. Engl. 49, 4771–4774 (2010).
Ellerbee, A. K. et al. Quantifying colorimetric assays in paper-based microfluidic devices by measuring the transmission of light through paper. Anal. Chem. 81, 8447–8452 (2009).
Martinez, A. W., Phillips, S. T., Wiley, B. J., Gupta, M. & Whitesides, G. M. FLASH: a rapid method for prototyping paper-based microfluidic devices. Lab. Chip. 8, 2146–2150 (2008).
Martinez, A. W. et al. Simple telemedicine for developing regions: camera phones and paper-based microfluidic devices for real-time, off-site diagnosis. Anal. Chem. 80, 3699–3707 (2008).
Annels, N. E. et al. Management of Epstein-Barr virus (EBV) reactivation after allogeneic stem cell transplantation by simultaneous analysis of EBV DNA load and EBV-specific T cell reconstitution. Clin. Infect. Dis. 42, 1743–1748 (2006).
Koelle, D. M. et al. Frequent detection of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) DNA in saliva of human immunodeficiency virus-infected men: clinical and immunologic correlates. J. Infect. Dis. 176, 94–102 (1997).
Bland, J. M. & Altman, D. G. Statistical methods for assessing agreement between two methods of clinical measurement. Lancet 1, 307–310 (1986).
Bland, J. M. & Altman, D. G. Measuring agreement in method comparison studies. Stat. Methods Med. Res. 8, 135–160 (1999).
Hong, J. et al. AC frequency characteristics of coplanar impedance sensors as design parameters. Lab Chip. 5, 270–279 (2005).
Rouet, F. et al. Transfer and evaluation of an automated, low-cost real-time reverse transcription-PCR test for diagnosis and monitoring of human immunodeficiency virus type 1 infection in a West African resource-limited setting. J. Clin. Microbiol 43, 2709–2717 (2005).
Deeks, S. G. et al. Towards an HIV cure: a global scientific strategy. Nat. Rev. Immunol. 12, 607–614 (2012).
World Health Organization. . Antiretroviral therapy in low- and middle-income countries by region. Available at: http://www.who.int/hiv/topics/treatment/data/en/. Date of access: 28/08/2014. (2011).
Marconi, V. C. et al. Cumulative viral load and virologic decay patterns after antiretroviral therapy in HIV-infected subjects influence CD4 recovery and AIDS. PLoS. One 6, e17956 (2011).
World Health Organization, March 2014 supplement to the 2013 consolidated guidelines on the use of antiretroviral drugs for treating and preventing HIV infection, Available at: . http://www.who.int/hiv/pub/guidelines/arv2013/arvs2013upplement_march2014/en/. (2014).
Stevens, G., Rekhviashvili, N., Scott, L. E., Gonin, R. & Stevens, W. Evaluation of two commercially available, inexpensive alternative assays used for assessing viral load in a cohort of human immunodeficiency virus type 1 subtype C-infected patients from South Africa. J. Clin. Microbiol. 43, 857–861 (2005).
Schupbach, J. Viral RNA and p24 antigen as markers of HIV disease and antiretroviral treatment success. Int. Arch. Allergy Immunol. 132, 196–209 (2003).
Labib, M., Shipman, P. O., Martic, S. & Kraatz, H. B. Towards an early diagnosis of HIV infection: an electrochemical approach for detection of HIV-1 reverse transcriptase enzyme. Analyst 136, 708–715 (2011).
van Oosterhout, J. J. G. et al. Diagnosis of antiretroviral therapy failure in Malawi: poor performance of clinical and immunological WHO criteria. Trop. Med. Int. Health 14, 856–861 (2009).
Mee, P., Fielding, K. L., Charalambous, S., Churchyard, G. J. & Grant, A. D. Evaluation of the WHO criteria for antiretroviral treatment failure among adults in South Africa. AIDS. 22, 1971–1977 (2008).
Calmy, A. et al. HIV viral load monitoring in resource-limited regions: optional or necessary? Clin. Infect. Dis. 44, 128–134 (2007).
Stevens, W. S., Scott, L. E. & Crowe, S. M. Quantifying HIV for monitoring antiretroviral therapy in resource-poor settings. J. Infect. Dis. 201, S16–26 (2010).
Tanriverdi, S., Chen, L. & Chen, S. A rapid and automated sample-to-result HIV load test for near-patient application. J. Infect. Dis. 201, S52–58 (2010).
Usdin, M., Guillerm, M. & Calmy, A. Patient needs and point-of-care requirements for HIV load testing in resource-limited settings. J. Infect. Dis. 201, S73–77 (2010).
World Health Organization. . Guidance on provider-initiated HIV testing and counselling in health facilities. Available at: http://whqlibdoc.who.int/publications/2007/9789241595568_eng.pdf. Date of access: 28/08/2014.
Keiser, O. et al. Switching to second-line antiretroviral therapy in resource-limited settings: comparison of programmes with and without viral load monitoring. AIDS. 23, 1867–1874 (2009).
Lynen, L., Van Griensven, J. & Elliott, J. Monitoring for treatment failure in patients on first-line antiretroviral treatment in resource-constrained settings. Curr. Opin. HIV. AIDS. 5, 1–5 (2010).
Balakrishnan, P. et al. Low-cost assays for monitoring HIV infected individuals in resource-limited settings. Indian J. Med. Res. 134, 823–834 (2011).
Damania, B., Lee, H. & Jung, J. U. Primate herpesviral oncogenes. Mol. Cells. 9, 345–349 (1999).
Chang, Y. et al. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266, 1865–1869 (1994).
Moore, P. S. & Chang, Y. Detection of herpesvirus-like DNA sequences in Kaposi's sarcoma in patients with and without HIV infection. New Engl. J. Med. 332, 1181–1185 (1995).
Chang, Y. et al. Kaposi's sarcoma-associated herpesvirus and Kaposi's sarcoma in Africa. Uganda Kaposi's Sarcoma Study Group. Arch. Intern. Med. 156, 202–204 (1996).
Said, J. W. et al. Primary effusion lymphoma in women: report of two cases of Kaposi's sarcoma herpes virus-associated effusion-based lymphoma in human immunodeficiency virus-negative women. Blood 88, 3124–3128 (1996).
Jones, D. et al. Primary-effusion lymphoma and Kaposi's sarcoma in a cardiac-transplant recipient. New Engl. J. Med. 339, 444–449 (1998).
Kikuta, H., Itakura, O., Taneichi, K. & Kohno, M. Tropism of human herpesvirus 8 for peripheral blood lymphocytes in patients with Castleman's disease. Brit. J. Haematol. 99, 790–793 (1997).
Soulier, J. et al. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood 86, 1276–1280 (1995).
Wabinga, H. R., Parkin, D. M., Wabwire-Mangen, F. & Mugerwa, J. W. Cancer in Kampala, Uganda, in 1989-91: changes in incidence in the era of AIDS. Int. J. Cancer 54, 26–36 (1993).
Athale, U. H., Patil, P. S., Chintu, C. & Elem, B. Influence of HIV epidemic on the incidence of Kaposi's sarcoma in Zambian children. J. Acquir. Immune. Defic. Syndr. Hum. Retrovirol. 8, 96–100 (1995).
Antman, K. & Chang, Y. Kaposi's sarcoma. N. Engl. J. Med. 342, 1027–1038 (2000).
Epstein, M. A., Achong, B. G. & Barr, Y. M. Virus Particles in Cultured Lymphoblasts from Burkitt's Lymphoma. Lancet 1, 702–703 (1964).
Johansson, B., Klein, G., Henle, W. & Henle, G. Epstein-Barr virus (EBV)-associated antibody patterns in malignant lymphoma and leukemia. I. Hodgkin's disease. Int. J. Cancer. 6, 450–462 (1970).
zur Hausen, H. et al. EBV DNA in biopsies of Burkitt tumours and anaplastic carcinomas of the nasopharynx. Nature 228, 1056–1058 (1970).
Ho, M. et al. Epstein-Barr virus infections and DNA hybridization studies in posttransplantation lymphoma and lymphoproliferative lesions: the role of primary infection. J. Infect. Dis. 152, 876–886 (1985).
Rickinson, A. B., E. . Kieff Epstein-Barr virus: In fields virology, 5th edition, edited by Fields BN, Knipe DM, Howley. Lippincott-Williams & Wilkins Publishers: Philadelphia, PA. 2655–2700 (2007).
Casper, C. et al. Frequent and asymptomatic oropharyngeal shedding of human herpesvirus 8 among immunocompetent men. J. Infect. Dis. 195, 30–36 (2007).
de Franca, T. R., de Araujo, R. A., Ribeiro, C. M. & Leao, J. C. Salivary shedding of HHV-8 in people infected or not by human immunodeficiency virus 1. J. Oral. Pathol. Med. 40, 97–102 (2011).
Mbulaiteye, S. M. et al. Detection of kaposi sarcoma-associated herpesvirus DNA in saliva and buffy-coat samples from children with sickle cell disease in Uganda. J. Infect. Dis. 190, 1382–1386 (2004).
Laney, A. S. et al. Human herpesvirus 8 presence and viral load are associated with the progression of AIDS-associated Kaposi's sarcoma. AIDS. 21, 1541–1545 (2007).
Quinlivan, E. B. et al. Elevated virus loads of Kaposi's sarcoma-associated human herpesvirus 8 predict Kaposi's sarcoma disease progression, but elevated levels of human immunodeficiency virus type 1 do not. J. Infect. Dis. 185, 1736–1744 (2002).
Rooney, C. M. et al. Early identification of Epstein-Barr virus-associated post-transplantation lymphoproliferative disease. Brit. J. haematol. 89, 98–103 (1995).
Riddler, S. A., Breinig, M. C. & McKnight, J. L. Increased levels of circulating Epstein-Barr virus (EBV)-infected lymphocytes and decreased EBV nuclear antigen antibody responses are associated with the development of posttransplant lymphoproliferative disease in solid-organ transplant recipients. Blood 84, 972–984 (1994).
Savoie A, P. C., Carpentier L,. Joncas J,. Alfieri C. . Direct correlation between the load of Epstein-Barr virus-infected lymphocytes in the peripheral blood of pediatric transplant recipients and risk of lymphoproliferative disease. Blood 83, 2715–2722 (1994).
Stevens, S. J. et al. Frequent monitoring of Epstein-Barr virus DNA load in unfractionated whole blood is essential for early detection of posttransplant lymphoproliferative disease in high-risk patients. Blood 97, 1165–1171 (2001).
Salmanzadeh, A. et al. Isolation of prostate tumor initiating cells (TICs) through their dielectrophoretic signature. Lab. Chip. 12, 182–189 (2012).
Salmanzadeh, A., Sano, M. B., Shafiee, H., Stremler, M. A. & Davalos, R. V. Isolation of rare cancer cells from blood cells using dielectrophoresis. Conf. Proc. IEEE. Eng. Med. Biol. Soc. 2012, 590–593 (2012).
Martinez, A. W., Phillips, S. T., Butte, M. J. & Whitesides, G. M. Patterned Paper as a Platform for Inexpensive, Low-Volume, Portable Bioassays. Angew. Chem. Int. Ed. 46, 2 (2007).
AS, D. et al. Top ten biotechnologies for improving health in developing countries. Nat. Genet. 32, 3 (2002).
Yager, P. et al. Microfluidic diagnostic technologies for global public health. Nature 442, 412–418 (2006).
Solomon, A. W. et al. A diagnostics platform for the integrated mapping, monitoring and surveillance of neglected tropical diseases: rationale and target product profiles. PLoS. Negl. Trop. Dis. 6, e1746 (2012).
Bruzewicz, D. A., Reches, M. & Whitesides, G. M. Low-cost printing of poly(dimethylsiloxane) barriers to define microchannels in paper. Anal. Chem. 80, 3387–3392 (2008).
Abe, K., Suzuki, K. & Citterio, D. Inkjet-printed microfluidic multianalyte chemical sensing paper. Anal. Chem. 80, 6928–6934 (2008).
Li, X., Tian, J., Nguyen, T. & Shen, W. Paper-based microfluidic devices by plasma treatment. Anal. Chem. 80, 9131–9134 (2008).
Lu, Y., Shi, W., Jiang, L., Qin, J. & Lin, B. Rapid prototyping of paper-based microfluidics with wax for low-cost, portable bioassay. Electrophoresis 30, 1497–1500 (2009).
Carrilho, E., Martinez, A. W. & Whitesides, G. M. Understanding wax printing: a simple micropatterning process for paper-based microfluidics. Anal. Chem. 81, 7091–7095 (2009).
Crombie, R. et al. Identification of a CD36-related thrombospondin 1-binding domain in HIV-1 envelope glycoprotein gp120: relationship to HIV-1-specific inhibitory factors in human saliva. J. Exp. Med. 187, 25–35 (1998).
Malamud, D., Davis, C., Berthold, P., Roth, E. & Friedman, H. Human submandibular saliva aggregates HIV. AIDS Res. Hum. Retrov. 9, 633–637 (1993).
Fox, P. C., Wolff, A., Yeh, C. K., Atkinson, J. C. & Baum, B. J. Saliva inhibits HIV-1 infectivity. J. Am. Dent. Assoc. 116, 635–637 (1988).
Alsip, G. R., Ench, Y., Sumaya, C. V. & Boswell, R. N. Increased Epstein-Barr virus DNA in oropharyngeal secretions from patients with AIDS, AIDS-related complex, or asymptomatic human immunodeficiency virus infections. J. Infect. Dis. 157, 1072–1076 (1988).
LaDuca, J. R. et al. Detection of human herpesvirus 8 DNA sequences in tissues and bodily fluids. J. Infect. Dis. 178, 1610–1615 (1998).
Pauk, J. et al. Mucosal Shedding of Human Herpesvirus 8 in Men. New Eng. J. Med. 343, 1369–1377 (2000).
Su, H. & Boackle, R. J. Interaction of the envelope glycoprotein of human immunodeficiency virus with C1q and fibronectin under conditions present in human saliva. Mol. Immunol. 28, 811–817 (1991).
Taraboletti, G., Roberts, D., Liotta, L. A. & Giavazzi, R. Platelet thrombospondin modulates endothelial cell adhesion, motility and growth: a potential angiogenesis regulatory factor. J. cell. biol. 111, 765–772 (1990).
Campbell, T. B. et al. Relationship of human herpesvirus 8 peripheral blood virus load and Kaposi's sarcoma clinical stage. AIDS. 14, 2109–2116 (2000).
Kalichman, S. C., Di Berto, G. & Eaton, L. Human immunodeficiency virus viral load in blood plasma and semen: review and implications of empirical findings. Sex. Transm. Dis. 35, 55–60 (2008).
Bourlet, T. et al. Compartmentalization of HIV-1 according to antiretroviral therapy: viral loads are correlated in blood and semen but poorly in blood and saliva. AIDS. 15, 284–285 (2001).
Moon, S. et al. Enumeration of CD4+ T-cells using a portable microchip count platform in Tanzanian HIV-infected patients. PLoS. ONE 6, e21409 (2011).
Myoung, J., Ganem, D. Generation of a doxycycline-inducible KSHV producer cell line of endothelial origin: Maintenance of tight latency with efficient reactivation upon induction. J. Virol. Methods 174, 12–21 (2011).
Miller, G., Shope, T., Lisco, H., Stitt, D. & Lipman, M. Epstein-Barr virus: transformation, cytopathic changes and viral antigens in squirrel monkey and marmoset leukocytes. Proc. Natl. Acad. Sci. U. S. A. 69, 383–387 (1972).
Acknowledgements
We would like to acknowledge NIH R01AI093282, NIH R01GM086382, NIH R01AI081534, NIH U54EB15408, NIH R21AI087107, NIH F32AI102590, NIH R01CA082036, the Brigham and Women’s Hospital Bright Futures Award, the Brigham and Women’s Hospital BRI Translatable Technologies and Care Innovation Grant and the Brigham and Women’s Hospital, Department of Medicine Innovation Evergreen Fund.
Author information
Authors and Affiliations
Contributions
H.S. and U.D. designed the study and the experiments; H.S., M.K.K., F.J., M.K., M.S., M. Y. and E.H. performed the experiments; E.H. and T.J.H. performed the HIV sequencing experiments; D.R.K. and K.M.K. supervised the virology aspect of the study and provided HIV, KSHV, EBV and E. coli samples; H.S., M.K.K., U.D. and F.J. wrote the paper; H.S. and U.D. supervised the research.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Shafiee, H., Kanakasabapathy, M., Juillard, F. et al. Printed Flexible Plastic Microchip for Viral Load Measurement through Quantitative Detection of Viruses in Plasma and Saliva. Sci Rep 5, 9919 (2015). https://doi.org/10.1038/srep09919
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep09919
This article is cited by
-
Gold nanoparticle/DNA-based nanobioconjugate for electrochemical detection of Zika virus
Microchimica Acta (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
