
Abstract
Accumulating evidence suggests that deposition of neurotoxic α-synuclein aggregates in the brain during the development of neurodegenerative diseases like Parkinson’s disease can be curbed by anti-aggregation strategies that either disrupt or eliminate toxic aggregates. Curcumin, a dietary polyphenol exhibits anti-amyloid activity but the use of this polyphenol is limited owing to its instability. As chemical modifications in curcumin confiscate this limitation, such efforts are intensively performed to discover molecules with similar but enhanced stability and superior properties. This study focuses on the inhibitory effect of two stable analogs of curcumin viz. curcumin pyrazole and curcumin isoxazole and their derivatives against α-synuclein aggregation, fibrillization and toxicity. Employing biochemical, biophysical and cell based assays we discovered that curcumin pyrazole (3) and its derivative N-(3-Nitrophenylpyrazole) curcumin (15) exhibit remarkable potency in not only arresting fibrillization and disrupting preformed fibrils but also preventing formation of A11 conformation in the protein that imparts toxic effects. Compounds 3 and 15 also decreased neurotoxicity associated with fast aggregating A53T mutant form of α-synuclein. These two analogues of curcumin described here may therefore be useful therapeutic inhibitors for the treatment of α-synuclein amyloidosis and toxicity in Parkinson’s disease and other synucleinopathies.
Similar content being viewed by others


Introduction
The aberrant self assembly and deposition of misfolded proteins is the leading cause for several conformational disorders like Alzheimer’s disease, multiple system atrophy and Parkinson’s disease.1,2 The term “α-synucleinopathy” refers to a specific group of such neurodegenerative diseases that exhibit abnormal aggregation and accumulation of α-synuclein, a pre-synaptic protein.3 Physiologically α-synuclein is a natively unfolded protein monomer regulated via lysosomal and proteosome degradation pathways.4,5 Nonetheless genetic and environmental stress factors disrupt normal physiological state of α-synuclein causing it to assemble and form toxic amyloid aggregates or highly ordered fibrils. Substitutional mutations (A30P, E46K and A53T) in α-synuclein and overexpression of the wild type α-synuclein (WtAS) due to gene multiplication are some of the identified genetic causes for this aberrant aggregation of α-synuclein.5
Parkinson’s disease (PD), a well-known α-synucleinopathy and most prevalent movement disorder in humans is characterized by the presence of α-synuclein aggregates as Lewy body inclusions in specific regions of the brain such as substantia nigra, thalamus and neocortex.6,7 Though numerous clinical and experimental studies have shown that soluble oligomeric and protofibrillar forms of α-synuclein are potentially neurotoxic, there are also reports stating neurodegenerative potency of fibrils via cell membrane permeabilization (Melki and Pieri, 2012).8,9,10 Molecules that inhibit α-synuclein fibrillization and stabilize it in a non-toxic state can therefore serve as therapeutic molecules for both prevention of accumulation of aggregated α-synuclein and maintenance of normal physiological concentrations of α-synuclein.11
A battery of evidences has shown that polyphenols along with their structural constraints are effective in amyloid inhibition.12,13 Curcumin (1,7-bis (4-hydroxy-3-methoxyphenyl)-1,6,-heptadiene-3,5-dione), a dietary polyphenol has been extensively studied and considered for treatment against neurodegenerative diseases not just for its renowned pharmacological activities and good safety profile but also for its unique ability to cross the blood brain barrier.14,15 Despite widely reported biological activities, the potential efficacy of curcumin is limited owing to its poor potency and bioavailability.16 A part of the rationale for this is its physical and metabolic instability which warrants its modification. Curcumin is a symmetric molecule with an α, β- unsaturated diketone moiety exhibiting keto-enol tautomerism. Attempts have been previously made by researchers to chemically modify curcumin to increase its potency. They include diketone and monoketone analogues of curcumin and knoevenagel condensates of curcumin.17,18 Our research group has previously reported that the pharmacological attributes of curcumin improve when 1, 3-dicarbonyl moiety of curcumin is replaced by isosteric isoxazoles and pyrazoles.19 Taking into account the structural relevance of this class of curcumin analogues, we hypothesized whether these compounds have any effect against aggregation and toxicity of α-synuclein.
In the current study, we therefore screened curcumin isoxazole (compound 2) and curcumin pyrazole (compound 3) for their inhibitory potency toward α-synuclein aggregation and compared the results with that of curcumin (compound 1). Employing several biophysical, imaging techniques, dot blot and cell based assays we have for the first time shown that curcumin pyrazole derivatives inhibit α-synuclein aggregation and reduce α-synuclein associated neurotoxicity. Our study also sheds light on the fact that compounds that inhibit fibrillization can have varying effect on neuronal cells.
RESULTS AND DISCUSSION
Chemistry
The preparation of isoxazole, pyrazole and substituted phenyl pyrazole derivatives of curcumin is illustrated in Figs. 1 and 2 as reported previously by us and others.20,21,22,23 The diketone moiety of curcumin was converted into its isoxazole with hydroxylamine hydrochloride by refluxing in glacial acetic acid for 6–8 h (Fig. 1). Pyrazole derivative of curcumin was synthesized by refluxing curcumin and hydrazine hydrate in glacial acetic acid for 6 h. Substituted curcumin pyrazoles were synthesized by the treatment of substituted phenyl hydrazine hydrochlorides with curcumin in glacial acetic acid under reflux condition for 8–24 h (Fig. 2). The structures of the synthesized curcumin analogues were confirmed by NMR spectroscopy and mass spectra. The purity of compounds was > 95% as determined by HPLC. The detailed synthesis of the compounds is shown in “Supplementary material 1” available online. Table 1 contains the list of compounds 1–19 and their structures. Hereafter curcumin analogues will be represented as numbered compounds.

Synthesis of Isoxazole and Pyrazole derivative of curcumin.

Synthesis of N-(substituted) phenylcurcumin pyrazole analogues.
Sequence of wild type and mutant α-synuclein cloned into pT7-7 vector was confirmed by sequencing. α-synuclein expressed in E.Coli BL21 cells was purified and the purity was confirmed by MALDI and Western blot (ESM_1 & ESM_2 “Supplementary Material 2”). ESM_3 (Supplementary Material 2) gives the visual confirmation of α-synuclein aggregation in 10 mM sodium phosphate buffer containing MgCl2 (10 mM) - pH 7.0 and 0.05% sodium azide over a period of 30 days at 37°C and 180 rpm.
Compound 3 is more potent inhibitor of wild type α-synuclein (WtAS) fibrillization than Compound 2
Since 2004 when curcumin was first reported to have anti-fibrillogenic property against protein misfolding and aggregation, extensive research work has been carried out to establish the mode of action of curcumin.24 However due to instability of curcumin in solution, chemical moiety responsible for such activities remains elusive. Nonetheless, efforts to explore the potency of stable curcumin analogues as inhibitors of α-synuclein amyloid have been lacking. Keeping in view the enormous beneficial effects of curcumin and its minimal toxicity, we have modified the chemical space around curcumin scaffold to synthesize stable curcumin isoxazoles and pyrazoles and studied the effect of these against α- synuclein aggregation and fibrillization.
As Thioflavin T (ThT) assay is a gold standard technique for quantification of amyloid fibrils, we monitored the change in dye fluorescence for screening the inhibitors against α-synuclein aggregation and fibrillization. However, ThT assay for detection of amyloid fibrils can be biased by the presence of polyphenolic compounds such as curcumin.25 Hence, concentrations of compounds were standardized at which there was negligible change in ThT fluorescence intensity. Moreover, since colored compounds and turbidity of the sample might quench ThT fluorescence and were likely to produce false positives, corrections for the inner filter effect were determined every time using equation (1) to rule out discrepancy caused by the above factors. Furthermore, to avoid any bias, we took into account the percent increase in ThT intensity instead of absolute intensities. Briefly, 3 mg/mL (~200 µM) of wild-type alpha synuclein (WtAS) was co-incubated with equimolar concentrations of compounds 2 and 3 for 30 days at 37°C with shaking at 180 rpm. The extent of aggregation was monitored by sampling at regular intervals. WtAS with compound 1 was used as a positive control while WtAS alone aggregated for 30 days served as a negative control. Fig. 3a (i) shows kinetics of WtAS aggregation in the absence and presence of compounds 1, 2 and 3 monitored by ThT binding assay. Percentage increase in ThT fluorescence for all the compounds were calculated using equation (2) . Our results showed that compound 3 significantly inhibited fibril formation as evidenced by reduced ThT fluorescence as compared to compound 2 (p = 0.0007) or 1 (p = 0.003) and the inhibitory potential of the compounds observed were of the order of compound 3 > compound 1 > compound 2. As pyrazole form of curcumin showed superior effect in ThT assay and curcumin isoxazole (compound 2) demonstrated significantly less effectiveness than curcumin (p = 0.02), we synthesized and investigated the potency of other pyrazole derivatives as well. Table 1 shows the percentage inhibition at 1:1 molar ratio of protein and compounds calculated using equation (3) . For a step-wise investigation, we analyzed whether analogues 3–19 possessed ability to (i) inhibit oligomerization or fibrillization; (ii) inhibit both oligomerization and fibrillization; (iii) disrupt existing oligomers and fibrils; (iv) modulate WtAS associated neurotoxicity.

(a) Time course of WtAS aggregation in the presence or absence of compounds 1–19 as monitored by ThT assay. WtAS (210 µM) was co-incubated with equimolar concentrations of curcumin (compound 1) or compounds 2–19 for 30 days. The ability of these compounds to prevent aggregation is shown as percentage increase in ThT fluorescence intensity. Aggregated samples were incubated with 50 µM of ThT for 30 min at 25°C. Samples were excited at 440 nm and emissions recorded at 480 nm. Results are the mean of three different experiments (n = 3) done in duplicate and the error bars show the standard deviations. # p < 0.005 compound 1 vs compound 3, compound 1 vs compound 15, $ p < 0.0001 compound 1 vs compound 6 (b) Percentage inhibition of aggregation of WtAS by compound 1–19 as monitored by ThT assay. WtAS (210 µM) was co-incubated with equimolar concentration of curcumin (compound 1) or compounds 2–19 for 30 days. The ability of these compounds to prevent aggregation is shown as percent inhibition of WtAS aggregation on day 30 as compared to WtAS alone. Aggregated samples were incubated with 50 µM of ThT for 30 min at 25 °C. Samples were excited at 440 nm and emissions recorded at 480 nm. Percent inhibition was calculated using appropriate equation. Results are the mean of three different experiments (n = 3) done in duplicate and the error bars show the standard deviations. # p < .005 compound 1 vs compound 3 and 15 and $ p < 0.0001 compound 1 vs compound 6.
Effect of compound 3 and its derivatives on kinetics of fibrillation and fibril morphology
Figs. 3a & 3b illustrate the effect of compounds against WtAS aggregation. ThT binding assays demonstrated that pyrazole derivatives 6 and 15 have significantly higher inhibitory potency than curcumin (Figs. 3a–ii & 3a–iv) with p values 0.00005 and 0.0005, respectively. The percentage inhibition of aggregation as measured from the ThT assays for compounds 9, 10 and 16 also showed inhibitory effect against α-synuclein aggregation; however they were less potent than curcumin (Fig. 3b). Out of 16 pyrazole derivatives, 11 did not exhibit any inhibitory effect against α-synuclein aggregation.
To validate the findings of ThT assay, the inhibition of WtAS aggregation was also confirmed by Congo red binding assay. A red shift in maxima of Congo red absorption spectrum from 490 nm to 540 nm indicates presence of increased cross β-sheet rich structures.26 We observed that this increased peak shift detected in WtAS alone aggregated control was prevented in WtAS samples containing compounds 3, 6 and 15 (Fig. 4a) corroborating the results of the ThT binding assay. To determine the inhibitor potency, different concentrations of compounds 3, 6 and 15 ranging from 1 µM to 500 µM were incubated with 210 µM of WtAS. The IC50 values were obtained by the dose response curves illustrated in Fig. 4b. 3 parameter sigmoidal fitting using equation (4) was done for the determination of IC50 values. While we obtained sigmoidal curves for compounds 1, 3 and 15, compound 6 showed a linear fitting. Linear dose response curves usually appear for compounds which are active at very low concentrations. Calculations showed that while compound 1 had IC50 value of 161.8 ± 1.24 µM, compounds 3 ((126.77 ± 1.10 µM) and 15 (89.13 ± 1.98 µM) inhibited WtAS at substantially less and substoichiometric concentrations relative to WtAS. Compound 6 exhibited maximum effect with an IC50 value of 9.21 ± 1.25 µM. The unusual fitting for compound 6 was because of its ability to inhibit WtAS aggregation even at very low concentrations.27

(a) Absorbance spectra of Congo Red of aggregated WtAS samples in the presence or absence of compounds 1, 3, 6 and 15. WtAS (210 µM) was co-incubated with equimolar concentration of compounds 1, 3, 6 and 15 for 30 days. The ability of these compounds to prevent aggregation was monitored by observing the peak shift from 490 nm to 540 nm. Samples were incubated with 50 µM Congo Red for 1 h at 37°C and Absorbance was scanned from 400 nm-600 nm using UV-Vis spectrophotometer. Results are mean of three independent experiments done in duplicate. (b) Dose response curves for determination of IC50 values of compounds 1, 3, 6 and 15 against WtAS aggregation. WtAS (210 µM) was co-incubated with various concentrations (1–500 µM) of compound 1, 3, 6 and 15 for 30 days. The ability of these compounds to prevent aggregation was monitored by ThT assay. Aggregated samples were incubated with 50 µM of ThT for 30 min at 25°C. Samples were excited at 440 nm and emissions recorded at 480 nm. The percentage of aggregation was calculated by measuring corrected ThT fluorescence post 30 days of incubation and was plotted against log of compound concentration. The 3-parameter sigmoidal fit gives the best fit and IC50 values were calculated from the graph. Results are mean of three independent experiments done in duplicate and the error bars show the standard deviation.
Though dye binding assays are used extensively for preliminary screening, other emerging complementary methods must also be used for assessing the fibrillization and inhibition phenomena. Hence, to substantiate the data obtained above WtAS protein was co-incubated with compounds 1, 3, 6 and 15 and their effect on α-synuclein aggregation were analyzed further using TEM and AFM which provides visual proof.
TEM photomicrographs of aggregated WtAS illustrated in Figs. 5a & 5b revealed that 30 day old WtAS fibrils were thin, long and in clusters. On the other hand α-synuclein co- incubated with compound 6 and 15 did not assume the fibrillar morphology and instead formed aggregates of smaller size and mixed morphology. WtAS co-incubated with compound 6 (Fig. 5e) formed ordered and compact aggregates whereas that incubated with compound 3 (Fig. 5d) and compound 15 (Fig. 5f) displayed amorphous and globular aggregates. The different patterns in which aggregates form on incubation with compounds 3, 6 and 15 suggested different ways by which these inhibitors act to prevent the fibril formation. AFM analysis also showed abundance of fully grown amyloid fibrils in control with heights of 1.9 ± 1.5 nm. However, as illustrated in Fig. 6 WtAS samples co-incubated with compounds 1, 3, 6 and 15 were mostly of oligomeric morphology having large differences in the morphology, distribution and heights. Oligomers of WtAS generated in the presence of compound 1 were comparatively larger in height (21.0 ± 2 nm) than compound 3 (6 ± 1 nm), compound 6 (5 ± 2 nm) and compound 15 (15 ± 3 nm) (Fig. 6). The average heights and diameter of aggregates in the presence of compounds 3, 6 and 15 were smaller in comparison to the aggregates formed in the presence of compound 1 as shown in histogram analysis in Fig. 6. The detailed size distribution of particles obtained from histogram analysis is also available online as Supplementary Material 3 as Table S1. The results of AFM clearly indicated that the compounds 3 and 6 and 15 interact with α-synuclein during the course of incubation and transform them into lower order oligomers as compared to the oligomers in the presence of compound 1. Collectively, above data and AFM results substantiated each other and indicated that these compounds modulated α-synuclein fibrillization.

Representative TEM images showing aggregate morphology of WtAS in the presence and absence of compounds 1, 3, 6 and 15.
WtAS (210 µM) was co-incubated with equimolar concentration of compound 1, 3, 6 and 15 for 30 days. The ability of these compounds to prevent aggregation was visualised by TEM. 3 µl aggregated samples were adsorbed on carbon coated formavar grids and negatively stained with 2% uranyl acetate for TEM imaging. Representative TEM images of WtAS (210 µM) (a) alone at day 0 (b) alone at day 30 (c) compound 1 (d) compound 3 (e) compound 6 (f) compound 15. Scale bar is 200 nm.

Representative AFM images showing aggregate morphology of WtAS in the presence of compounds 1, 3, 6 and 15.
WtAS (210 µM) was co-incubated with equimolar concentration of compound 1, 3, 6 and 15 for 30 days. The ability of these compounds to prevent aggregation was visualised by AFM. Representative AFM topographic images of ultrastructure of 50X diluted WtAS (210 µM) aggregates (a) alone at day 30 (b) compound 1 (c) compound 3 (d) compound 6 (e) compound 15. Right panel shows the histogram analysis depicting size distribution.
Characterization of binding of compound 3 and its derivatives to WtAS by employing compound fluorescence
In order to evaluate the binding of compounds 1, 3, 6 and 15 to WtAS, all the compounds were excited at their absorption maxima and fluorescence emission spectra were recorded. Fluorescence spectra of WtAS co-incubated with compounds 1, 3, 6 and 15 showed that all these analogues exhibit fluorescence emission at much lower wavelength as compared to curcumin (compound 1) (Fig. 7). All the compounds showed a blue-shift in the emission maxima producing a characteristic peak at 400 nm because of the substitutions in the diketone moiety which hinders extended conjugation.28

Fluorescence spectra of compounds 1, 3, 6 and 15 in the presence of equimolar concentration of WtAS.
Samples of of WtAS (20 µM) containing compounds 1, 3, 6 and 15 were incubated for 15 min and excited at their wavelength maxima; 427 nm for curcumin and 330 nm for compounds 3, 6 and 15. Results are mean of three different experiments done in duplicate.
Characterization of binding of compound 3 and its derivatives to WtAS by employing intrinsic tyrosine fluorescence
Alpha-synuclein is a Class-A protein as it does not contain any tryptophan residue.29 Any fluorescence exhibited by α-synuclein at emission maxima of 303 nm, pH 7.0 is attributed to the presence of four tyrosine residues in it.30 Any conformational change in the protein may increase or decrease the intrinsic fluorescence intensity. Here, spectral changes in tyrosine fluorescence of WtAS were monitored after co-incubation of compounds 1, 3, 6 and 15. A substantial quenching of the fluorescence intensity (Fig. 8) of WtAS was observed with maximal decrease observed for compound 6. The decrease in intrinsic fluorescence intensity suggests that compounds 1, 3, 6 and 15 interacted with WtAS resulting in conformational change of the protein. It also gave an indication that compound 6 causes maximal change in the protein conformation.30

Intrinsic fluorescence spectra of WtAS induced by the binding of compounds 1, 3, 6 and 15.
WtAS (70 µM) was incubated in the presence and absence of equimolar concentrations of compounds 1, 3, 6 and 15 at 25°C pH 7.0 for 1 hr. Binding of compounds to protein was inferred by monitoring the intrinsic fluorescence of WtAS contributed solely by tyrosine residues. Samples were excited at 280 nm and emission was recorded in the range of 285–500 nm. Results are mean of three different experiments done in duplicate.
Effect of curcumin pyrazole and its derivatives on existing and pre-formed α-synuclein fibrils
For all aberrant protein aggregation disorders, an important and alternative therapeutic strategy is to either halt the process of aggregation or altogether reverse the process in order to reduce the burden of amyloid in the diseased brain. A few molecules are known to reverse aggregation of α-synuclein. Herein we tested whether compounds 3, 6 and 15 possessed any ability to disassemble preformed aggregates. To monitor the effect of compounds 3, 6 and 15 on existing WtAS aggregates; we added these compounds in reaction mixtures on days 15 and 30 post initiation of protein aggregation and further left it to aggregatefor another 30 days. ThT and Congo Red binding assays were then done on days 45 (15 + 30) and 60 (30 + 30), . Figs. 9a and 10a illustrate the extent of amyloid formation as evidenced by ThT binding. Statistical analysis showed that when compared to WtAS alone, all the compounds showed significant inhibition on day 45. The order of inhibition of elongation was compound 6 > compound 1 > compound 15 > compound 3 with p values 0.0002, 0.0003, 0.0009 and 0.011 for compounds 6, 1, 15 and 3 respectively (Fig. 9a). Disruption analysis with ThT on day 60 followed the order: compound 3 > compound 6 > compound 15 > compound 1 (Fig. 10a). All the compounds caused significant disruption with p values 0.0006, 0.0007 and 0.002 for compounds 3, 6, ad 15 as compared to WtAS aggregated alone except curcumin (p = 0.2). From this it was found that compound 3 which showed least effect on day 45 exhibited maximum effect on day 60. These data indicated that compounds 3 and 6 have highest affinity for binding sites on fully grown fibrils and intermediate oligomers, respectively. Compounds 6 and 15 did not show any particular difference in the effect on day 15 and day 30 aggregates as evidenced by ThT assay. It also showed that the effect of compound 1 (curcumin) can wear out upon incubation. Again when Congo red spectrum was recorded for the same samples, we found that all the compounds lead to a reversal of peak shift caused by the amyloid (Figs. 9b & 10b).

(a) Comparative bar diagram showing the effect of compounds 1, 3, 6 and 15 on 15 days aggregated WtAS by ThT assay. WtAS (210 µM) samples were aggregated alone for 45 days or equimolar concentrations of compounds 1, 3, 6 and 15 were added at day 15 and further co-incubated for 30 days. The ability of these compounds to prevent further aggregation was monitored by ThT assay. Aggregated samples were incubated with 50 µM of ThT for 30 min at 25 °C. The samples were excited at 440 nm and emissions recorded at 480 nm. Percentage increase in aggregation was calculated by measuring corrected ThT fluorescence intensity. Results are the mean of three different experiments done in duplicate and the error bars show the standard deviations. * p < 0.05 control vs compound 3, # p < .005 control vs compounds 1 & 15 and $ p < 0.0001 control vs compound 6. (b) Absorbance spectra of Congo Red of aggregated WtAS samples in the presence or absence of compounds 1, 3, 6 and 15 on 15 days aggregated WtAS. WtAS (210 µM) samples were aggregated alone for 45 days or equimolar concentrations of compounds 1, 3, 6 and 15 were added at day 15 and were further co-incubated for 30 days. The ability of these compounds to prevent further aggregation was monitored by Congo red binding assay. Samples were incubated with 50 µM Congo Red for 1 h at 37°C and Absorbance was scanned from 400 nm–600 nm using UV-Vis spectrophotometer. Results are mean of three independent experiments done in duplicate.

(a) Comparative bar diagram showing the effect of compounds 1, 3, 6 and 15 on 30 days aggregated WtAS by ThT assay. WtAS (210 µM) samples were aggregated alone for 60 days or equimolar concentrations of compounds 1, 3, 6 and 15 were added at day 30 and further co-incubated for 30 days. The ability of these compounds to prevent further aggregation was monitored by ThT assay. Aggregated samples were incubated with 50 µM of ThT for 30 min at 25°C. The samples were excited at 440 nm and emissions recorded at 480 nm. Percentage increase in aggregation was calculated by measuring corrected ThT fluorescence intensity. Results are the mean of three different experiments (n = 3) done in duplicate and the error bars show the standard deviations. # p < .005 control vs compounds 3, 6 & 15. (b) Absorbance spectra of Congo Red of aggregated WtAS samples in the presence or absence of compounds 1, 3, 6 and 15 on 30 days aggregated WtAS. WtAS (210 µM) samples were aggregated alone for 60 days or equimolar concentrations of compounds 1, 3, 6 and 15 were added at day 30 and were further co-incubated for 30 days. The ability of these compounds to prevent further aggregation was monitored by Congo red binding assay. Samples were incubated with 50 µM Congo Red for 1 h at 37°C and Absorbance was scanned from 400 nm–600 nm using UV-Vis spectrophotometer. Results are mean of three independent experiments done in duplicate.
The results implied that compounds 3, 6 and 15 were potent enough to disaggregate mature preformed fibrils and even to slow down further elongation of shorter fibrils even when added at a stage where aggregation is rampant or towards completion. It was also seen that among all compound 3 was best at disrupting fibrils while compound 6 was a better inhibitor of fibril elongation.
AFM analysis was also carried out to visualize the extent of disruption of 60 day WtAS samples aggregated alone or with compounds 1, 3, 6 and 15 added on day 30. As illustrated in Fig. 11 compounds 1, 3, 6 and 15 were able to disrupt the preformed fibrils with varying potency. The analysis of samples with AFM showed that efficiency of disruption followed the order; compound 6 ≥ compound 3 > compound 15 > compound 1. WtAS fibrils co-incubated in the presence of compound 1 curcumin) were broken down into smaller fragments with fibrillar morphology whereas the presence of compounds 3 & 6 led to complete disruption of the fibrils into smaller oligomers. However, in case of compound 15, an almost homogenous population of higher order oligomeric structures with unique morphology alongwith small oligomers was found. The results again reconfirmed that compounds have different mechanism of fibril disruption and need independent investigation.

Representative AFM images showing morphology of species generated when preformed WtAS fibrils incubated in the presence or absence of compounds 1, 3, 6 and 15 for 30 days.
WtAS (210 µM) samples were aggregated alone for 60 days or equimolar concentrations of compounds 1, 3, 6 and 15 were added at day 30 and were further co-incubated for 30 days. The ability of these compounds to disrupt preformed fibrils was visualised by AFM. Representative AFM topographic images of ultrastructure of 100X diluted WtAS (210 µM) aggregates (a) alone at day 60 (b) compound 1 (c) compound 3 (d) compound 6 (e) compound 15. Right panel shows the histogram analysis depicting size distribution.
Structure activity relationships
When 1, 3-dicarbonyl group in curcumin was substituted by isosteric heterocycles like isoxazole and pyrazole, an alteration in the inhibitory effect of curcumin against α-synuclein aggregation was observed with curcumin isoxazole (compound 2) exhibiting less inhibitory effect than curcumin and pyrazole-curcumin (compound 3) showing better inhibitory effect as compared to curcumin. This was the rationale for the synthesis of phenylpyrazole (compound 4) and 15 more pyrazole derivatives (compounds 5–19). The effects of various substituents in the phenyl group were investigated in order to find out their structure activity relationship. Addition of phenyl ring in pyrazole moiety (compound 4) did not modulate the activity of the parent compound any further. However incorporation of fluoro group in phenylpyrazole curcumin ring at meta position (compound 6) raised the anti-aggregation potential of the compounds twice as compared to curcumin. Nonetheless substitution of same fluoro group in phenylpyrazole curcumin at ortho (compound 5) and para (compound 7) positions abolished the inhibitory activity completely. Incorporation of electron withdrawing nitro group in the N-phenylpyrazole curcumin moiety at meta position (3-nitro Phenylpyrazole curcumin, compound 15) increased the anti-aggregation potential of curcumin almost equipotent to curcumin pyrazole but when the nitro group was substituted at ortho position, the anti-amyloid activity of 2-nitro-phenylpyrazole curcumin (compound 14) drastically decreased. It was also found that incorporation of electron donating group like o-Tolyl (compound 12) and m-Tolyl (compound 13) in pyrazole derivative of curcumin abrogated the anti-aggregation potential of curcumin pyrazole. Remarkably, even introduction of electron donating group (−OCH3) at para position on phenylpyrazole curcumin (compound 16) imparted a better anti-aggregation potential than curcumin but less when compared to curcumin pyrazole.
Addition of carboxyl group (compound 17), benzyl group (compound 18) or hydroxyl ethylene group (compound 19) in phenylpyrazole curcumin compound also decreased the anti-aggregation potential of pyrazole curcumin drastically. Interestingly, compounds 2-chloro-phenylpyrazole curcumin (compound 8) and 4-bromo-phenyl pyrazole curcumin (compound 11) were observed to aggravate α-synuclein fibril formation while meta chloro (compound 9) and ortho-para dichloro (compound 10) substituted phenylpyrazole compounds showed reduced inhibitory activity than pyrazole curcumin implying that substitution of certain groups can modulate the normal aggregation of α-synuclein.
Overall, our data revealed the efficacy of the compounds in the order of Compound 6 > Compound 15 > Compound 3 > Compound 16 > Compound 1
We presume that pharmacological effect of compound 6 (N-(3-Fluorophenylpyrazole) curcumin) as an inhibitor of fibril elongation could be attributed to the presence of a fluorine group at the 3rd position of phenyl pyrazole and not elsewhere in the ring as when placed at the 4th position of phenyl pyrazole (compound 7), this beneficial effect was not observed. Our findings are strengthened by previous reports which show that small molecules have the potential to prevent aggregation as well as fibrillization. Many compounds such as Congo Red (a potent amyloid inhibitor), Chrysamine G (a blood brain permeable molecule) and RS-0406 (novel β- sheet breaker) which are structurally similar to curcumin have also been shown to prevent protein aggregation.31,32,33
As the overall structure activity relationship showed that a minor alteration in the phenylpyrazole curcumin compounds can have a pronounced effect on α-synuclein aggregation, the present study opens a new era for exploring suitably designed curcumin derivatives as potential α-synuclein inhibitor.
Effect of curcumin pyrazoles on α-synuclein oligomer toxicity
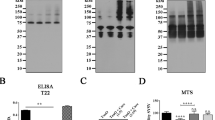
For a compound to qualify as a therapeutic molecule, it should not only inhibit aggregation but also modulate the toxicity of the oligomeric species produced during the fibrillization. Previous reports have shown that curcumin not only alters the structure of oligomeric species but also modulates and renders them less pathogenic.34 Studies have shown that depending upon the extent of proteinase K (PK) digestion, the toxicity of oligomers may vary. Oligomers resistant to PK digestion are mostly stable and generate large amount of toxic reactive oxygen species.35 To test whether WtAS aggregates containing curcumin pyrazole and selected derivatives were PK resistant, we incubated WtAS for 30 days in the presence of compounds 1, 3, 6, 15 followed by Proteinase-K digestion. Coomassie staining of the digested samples run on SDS-PAGE showed marked reduction of PK resistant core in the presence of compounds 3, 6 and 15 than with compound 1. Results clearly suggested that the oligomers formed in the presence of compounds 1, 3, 6, 15 are susceptible to degradation by PK (Fig. 12). Nonetheless, susceptibility to PK digestion does not give any idea regarding the nature of the oligomers because even PK sensitive oligomers may contribute to toxicity.36

Representative 12% SDS-PAGE gel image showing Proteinase-K digestion products of WtAS aggregated alone or in the presence of compounds 1, 3, 6 and 15.
WtAS (210 µM) were aggregated alone or co-incubated with equimolar concentration of compound 1, 3, 6 and 15 for 30 days. 20 µl WtAS samples were subjected to digestion by 50 µg/ml of Proteinase-K for 1 h at 37°C and boiled at 100°C for 5 min after adding SDS sample buffer. Lane 1-undigested, Lane 2–6 digested samples of only WtAS with 1, 3, 6 and 15.
To further ascertain the toxicity of the species, we used a conformation dependent antibody A11 which binds specifically to the toxic epitopes of all the majorly aggregating proteins such as Aβ, tau and synuclein.37 Dot blot analysis showed that protein aggregates containing curcumin from day 0 exhibited slight reactivity against A11 while compounds 3 and 15 totally suppressed the formation of this toxic epitope at the end of 30 days (Fig. 13a). Strikingly, compound 6 did not modulate A11 conformation in α-synuclein at all. We presume that the differences in immunoreactivity of the protein end-products containing curcumin and its pyrazole derivatives could be due to different mechanisms of oligomerization in the presence of different compounds.

Representative image of A11 dot blot showing effect of compounds 1, 3, 6 and 15 on WtAS A11 epitope formation.
(a) WtAS (210 µM) aggregated alone or co-incubated with equimolar concentration of compound 1, 3, 6 and 15 for 30 days (b) WtAS (210 µM) samples aggregated alone for 60 days or with equimolar concentrations of compounds 1, 3, 6 and 15 added at day 30 and further co-incubated for 30 days. For dot blot analysis 3 μL of WtAS samples were spotted on a nitrocellulose membrane and probed with A11 antibody.
Next the compounds 3, 6 and 15 were evaluated for their ability to modulate the toxicity of preformed aggregates. Compounds 3, 6 and 15 were added to day 30 WtAS aggregated protein and incubated for another 30 days. As evident from data compounds 3, 6 and 15 showed superior effects in disrupting preformed fibrils. No change in A11 reactivity was seen in compound 6 as compared with α-synuclein control (Fig. 13b). The results indicated that though compound 6 prevented fibrillization of WtAS as well as disrupted the preformed fibril efficiently, it yielded oligomers that were toxic in nature. However, cell based experiments were required to confirm the same
To further investigate whether the end products of WtAS incubated with compound 3, 6 and 15 were toxic or not, we employed SHSY5Y neuroblastoma cell line to evaluate their cytotoxicity. SH-SY5Y, an adrenergic cell line routinely used to study neuronal functions expresses dopaminergic markers and hence is used for studying PD. SH-SY5Y cells incubated for 24 hrs with either 30 day WtAS aggregates alone or those containing compounds 1, 3, 6 and 15 were assessed for cell viability using MTT assay. MTT assay does not directly measure cell death; rather it gives a measure of the ability of cells to reduce MTT dye producing coloured end products. The amount of colour produced as compared to the control when quantified gives an indication of cell viability. The percentage cell viability was calculated using equation (5) . Results showed that protein samples containing compounds 3 and 15 conferred no toxicity in neuronal cells whilst compound 6 displayed considerably high neuronal toxicity (Fig. 14). Vehicle employed did not confer any cell toxicity.

Comparative bar diagram showing MTT reduction by oligomers of WtAS alone or co-incubated with compounds 1, 3, 6 and 15 in neuronal cultures.
WtAS (210 µM) was aggregated alone or co-incubated with equimolar concentration of compounds 1, 3, 6 and 15 for 30 days. SH-SY5Y cells were co-incubated with 2 µl of above samples for 24 h followed by MTT addition. The products of MTT reduction were dissolved and colour was read at 570 nm. Percentage MTT reduction was calculated. Results are the mean of three different experiments done in duplicates and error bars show standard deviation. $ p < 0.0001 vehicle control vs WtAs & WtAS + compound 6.
Thus both the results of A11 immunoblot assay and MTT corroborated and gave a clear indication that the oligomers of WtAS formed with incubation of compound 6 were toxic by nature. Nonetheless, compound 6 alone did not impart any cytotoxicity. Toxicity was solely attributed to the oligomers the generated by the compound during the course of WtAS aggregation and disruption of fibers.
Curcumin pyrazole and its derivatives effectively inhibit the fibrillization of faster aggregating A53T mutant α-synuclein
Reports on PD have suggested that α-synuclein undergoes aberrant aggregation by PD-linked mutations such as A30P, A53T and E46K. A53T mutant α-synuclein has the tendency to form oligomers and fibrils at a faster rate than wild type leading to early onset forms of PD.38,39 Herein we tested the inhibitory efficiency of compound 3, 6 and 15 on the aggregation of A53T mutant synuclein. TEM images of A53T mutant α-synuclein incubated with compounds 3, 6 and 15 (Fig. 15a) for 30 days depict a clear inhibition of the fibril formation suggesting that the curcumin pyrazole (compound 3) and its derivatives compounds 6 and 15 not only inhibit the aggregation of wild type protein but also inhibit aggregation of deleterious mutant forms of the protein. It was also observed that compound 6 aided formation of dense aggregates as compared to compounds 3 and 15 which showed diffused aggregates in a pattern as seen when co-incubated with WtAS. Dot blot assays conducted on these samples showed that both compounds 3 and 15 prevented the formation of toxic species when added at day 0 (Fig. 15b) and compound 3 was very effective in completely preventing the A11 epitope even when incubated with 30 days fibrils for another 30 days. (Fig. 15b). Though compound 15 was not able to modulate the toxicity of mutant α-synuclein completely on addition at day 30, it has potency to alleviate the toxicity on addition at day 15 post initiation of aggregation. Both the compounds 3 and 15 were superior to curcumin in inhibition of mutant α-synuclein fibrillization and reduction in toxicity. In conjunction with the data obtained from aggregation experiments, compound 6, unlike other derivatives neither prevented nor reduced A11 conformation (Fig. 15b) even though it effectively inhibited α-synuclein fibrillization.

Representative TEM micrographs and A11 dot blot images showing aggregate morphology and toxicity of A53T mutant α-synuclein in the absence or presence compounds 1, 3, 6 and 15.
(a) TEM images of A53T mutant α-synuclein (210 µM) aggregated for 30 days (A) alone or in the presence of equimolar concentrations of (B) compound 1 (C) compound 3 (D) compound 6 (E) compound 15. Scale bar is 200 nm. (b) A11 dot blots showing effect of compounds 1, 3, 6 and 15 on A53T mutant α-synuclein A11 epitope formation. A53T mutant α-synuclein samples (210 µM) were aggregated alone or co-incubated with equimolar concentration of compounds 1, 3, 6 and 15 for 30 days or 60 days. Compounds were added on day 0 and day 30 respectively.
The results of our study highlight different means by which curcumin and curcumin pyrazole derivatives affect α-synuclein aggregation and fibrillization. Previous studies have indicated that aggregation of α-synuclein to oligomers and fibrils is mainly related to its structure and peptide sequence. There are 3 main regions in α-synuclein which include (i) N-terminal peptide sequence (residues 1–65), which contains the imperfect 11-residue repeats that enable the protein to achieve amphipathic α-helical conformation upon interaction with negatively charged phospholipids (ii) non-Aβ component of amyloid (NAC) or the hydrophobic core (residues 66-95) which confers β-sheet conformation and self-aggregation propensity under pathological conditions and (iii) the carboxy terminus (residues 96-140), a negatively charged region critical for self-assembly and interaction with other proteins and small molecules. In silico studies have shown that curcumin preferentially associates with aliphatic residues like alanine in fast aggregating Aβ peptide.40 Lapidus et al. have proposed that presence of a number of aliphatic residues like Alanine and Valine in the central NAC region of α-synuclein facilitate the binding of more than one curcumin to the pathogenic protein, allowing faster reconfiguration, preventing interactions between monomers and slowing the overall aggregation process.41 Thus, amino acids with aliphatic side chains in α-synuclein could also be the binding sites for compounds 3 and 15 and this may be a mechanism by which these compounds reduce α-synuclein aggregation and toxicity. Moreover, these compounds have even a higher degree of structural resemblance than curcumin to the novel β- sheet breaker small molecule N,N’-bis (3-hydroxyphenyl)pyridazine-3,6 diamine (RS-0406).32 Alternatively, it is also possible that these compounds can also directly bind to fibrillar assembly in a way similar to but more efficiently than curcumin and disrupt preformed fibrils.
Nevertheless, the case with compound 6 appears different. Turbidity measurements at 360 nm were made of α-synuclein aggregated for 30 days in the presence of compounds 1, 3, 6, 15 to characterize the oligomers formed (Fig. 16). Compound 6 considerably reduced turbidity as compared to WTAS alone (p = 0.0048) which indicated that end-products of compound 6 reactions are mostly soluble in nature. From AFM, TEM and dot blot analysis we deduced that though compound 6 inhibited α-synuclein fibrillization and disrupted existing fibrils, it favoured the formation of pathophysiological A11 epitope of aggregated alpha-synuclein. Compound 6 broke down α-synuclein effectively but the oligomeric by-products of the reaction were toxic. The results indicate that although compounds which appear to reduce cross beta-sheet content of α-synuclein, reducing aggregation and disrupting existing fibrils, it may not impart beneficial effects owing to the retention of toxicity. Our results are in line with those reported earlier that soluble oligomers and smaller fibrils are toxic by nature. At the same time inhibition of formation of oligomers of one kind can lead to formation and stabilization of an altogether different class of oligomers.8 Moreover, neither is each inhibition beneficial nor are all oligomers toxic.42 This also reinforces the fact that fibrillization is independent and distinct from oligomerization. For example- Compound 6 inhibits both fibrillization and insoluble oligomerisation but promotes formation of soluble, prefibrillar, toxic on-pathway oligomers while the presence of compounds 3 and 15 inhibit formation of fibrils but promote stabilization of some off-pathway insoluble aggregates which lack toxicity as suggested by A11 reactivity.

Comparative bar diagram showing changes in turbidity of aggregated WtAS samples in the presence and absence of compounds 1, 3, 6 and 15.
WtAS (210 µM) was co-incubated with equimolar concentration of compound 1, 3, 6 and 15 for 30 days. The changes in turbidity were monitored by turbidity assay. 5 µl samples of WtAS samples were added to 145 µl of aggregation buffer and absorbance at 360 nm was recorded using UV-Vis spectrophotometer. Increase in turbidity was calculated by subtracting the turbidity at day 0. Results are mean of three independent experiments done in duplicate. $ p < 0.0001 control vs compound 6.
To conclude, we report that compound 3 and its derivate, compound 15(N-(3-Nitrophenylpyrazole) Curcumin are potent therapeutic molecules that inhibit the progression of α-synuclein aggregation. These two compounds are anti-fibrillization agents that modulate α-synuclein associated neurotoxicity. The compounds may therefore have both prophylactic and therapeutic effects in the PD brain milieu.
Our data also suggests that compounds like compound 6 (N-(3-Fluoro phenylpyrazole) Curcumin which seem therapeutic via conventional biophysical and imaging techniques do not impart any beneficial effects in reducing cytotoxicity. The results indicate that conventional dye binding assays like ThT and Congo Red along with microscopic imaging should be done in conjunction with cell-based assays to determine the nature of the compound under study which again underscores the importance of our work. Any strategy to discover new anti-amyloidogenic molecules should take into account the toxicity of the resulting oligomers. The future prospects of the study include investigating the modulatory role of not only compounds 3 and 15 but also compound 6 in disease progression in α-synuclein overexpressing transgenic mice models. The use of compounds 6 and 3 in mechanistic studies may enable a better understanding of the pathogenesis of α-synuclein in the progression of PD and other synucleinopathies. A summary of the results is shown in Table 2 with the detailed graphical abstract of the work in Fig. 17

Graphical abstract illustrating the effect of Compound 3 (curcumin pyrazole), Compound 15 (N-3-Nitrophenylpyrazole curcumin) and Compound 6 (N-3-Fluoro phenylpyrazole curcumin) on α-synuclein aggregation and fibrillization.
Methods
Materials
All solvents used for the synthesis of curcumin and its analogues were either of spectral grades or distilled prior to use. Solvents and other reagents for synthesis were procured from Sigma Aldrich, USA unless otherwise mentioned. Chemicals used for bacterial cell culture and mammalian cell cultures were obtained from Himedia, India and Gibco, USA respectively. Antibodies against α-synuclein were procured from Santa cruz Biotechnology Inc, USA while A11 antibody was obtained from Invitrogen, CA, USA. All other chemicals used were from Sigma Aldrich, USA.
Preparation of Inhibitors
Stock solutions (1 mM) of the compounds were prepared in DMSO. Final concentration of DMSO in the reaction mixture including control was 20%.
Expression and purification of α-synuclein
α-synuclein, (Wild-type and A53T mutant) was expressed in Escherichia coli using plasmid pT7-7 encoding for the protein (were a kind gift from Dr. Rajiv Bhat, School of Biotechnology, Jawaharlal Nehru University, New Delhi, India). Following transformations, BL21 cells were grown in LB in the presence of ampicillin (100 µg /mL). Cells were induced with 1 mM IPTG and harvested four hour post-induction by centrifugation at 5,000 rpm for 15 min. The cell pellet was re-suspended in buffer (50 mM Tris-HCl (pH 8.0) containing 10 mM EDTA and 15 mM NaCl) with protease inhibitor cocktail and lysed by vigorous vortexing. The cell suspension was boiled for 30 min, followed by centrifugation for 30 min at 14,000 rpm and 4 °C. 10% streptomycin sulphate (136 µL/mL) was added to the supernatant followed by glacial acetic acid. After another 10 min of centrifugation at 14,000 rpm, equal volume of saturated ammonium sulphate was added to the supernatant. The solution was centrifuged again and now the pellet was resuspended in 1:1 mixture of chilled 100 mM ammonium acetate and absolute ethanol. The pellet obtained after centrifugation at 14,000 rpm once more was washed with absolute ethanol till it was white. Final pellet obtained was air-dried to remove traces of ethanol completely and was resuspended in the required 10 mM sodium phosphate (pH 7.0) buffer with 0.05% sodium azide. Purity of protein was confirmed by western blot and mass-spectrometry. To eliminate batch to batch variations, several preparations were pooled, so that a single homogenous preparation was used throughout the study. Protein concentration was estimated using Nano Drop (Thermo 1000) from the absorbance at 280 nm using molecular weight 14.46 kDa and an extinction coefficient of 5960 M−1cm−1. Small aliquots of the protein were stored at −20°C.
Aggregation Assays
3 mg/mL (210 µM) of α-synuclein was aggregated in aggregation buffer (10 mM sodium phosphate (pH 7.0) buffer containing 10 mM MgCl2 and 0.05% sodium azide) with constant shaking at 180 rpm at 37°C in the absence or presence of 210 µM of compounds. 20% of DMSO was added to all the negative control reaction mixtures. Aliquots were withdrawn at different time points for various biophysical assays.
Thioflavin T (ThT) Binding Assay
A 5 mM aqueous solution of Thioflavin T was prepared and filtered through 0.2 μm polyether sulfone filter. 2 μL of 210 μM α-synuclein sample solution was diluted in 1 mL of 50 μM ThT (dissolved in Glycine-NaOH buffer, pH 8.0) and incubated for 15 min at 25°C in the dark. The resulting ThT fluorescence intensities of samples were measured at an emission wavelength of 480 nm using an excitation wavelength of 440 nm and 5 nm slit width using a Jobin Vyon Horiba Fluoromax-4 spectrofluorimeter. The inner filter effect was taken into account using the equation (1)

Where Fc is the corrected fluorescence and F is apparent fluorescence, Aex and Aem are the absorbances of the samples at excitation and emission wavelengths respectively. To account for differences in protein samples, all the measurements were normalised by subtracting the day 0 values from all the measured values. Percent change in fluorescence intensity (Fp) was calculated using the equation (2)

Where Ft is fluorescence at a specific time point and F0 is fluorescence on day 0. All experiments were done in triplicate.
Percent inhibition was calculated using equation (3)

Where Fx is percent inhibition, Fs is percent increase in fluorescence intensity in the presence of compounds and Fc is the percent increase in fluorescence intensity of synuclein alone.
IC50 of compounds were calculated from the dose response curve using 3 parameter sigmoidal fitting as shown in equation (4)

where X is the log of concentration and X0 is the log of concentration at 50% inhibition.
Congo Red Absorbance Assay
A 50 µM solution of Congo Red (CR) was prepared in buffer (5 mM potassium phosphate, 150 mM NaCl, pH 7.4) and filtered through 0.2 µM syringe filter. 10 µL of protein samples were added to 190 µL of Congo Red and incubated for 1 h at 37°C. Absorbance data were recorded as a spectrum from 400-600 nm on Shimadzu UV-VIS spectrophotometer.
Transmission Electron Microscopy (TEM)
A 3 μL α-synuclein sample was adsorbed onto a Carbon coated Formvar mesh grid for 2 min. The grids were then negatively stained with 2% Uranyl acetate for 1 min and blotted dry. Transmission electron microscopy images were collected with a CM10 Philips operated at 80 kV.
Atomic Force Microscopy (AFM)
Protein samples were mixed gently to resuspend any aggregates. AFM experiments were carried out using the direct surface adsorption on freshly cleaved mica. For visualization samples were diluted 50 folds with nano-pure water. 5 μL of obtained solution was immediately deposited onto the freshly cleaved mica surface and left for 2 min. After that, each sample was rinsed with nano-pure water and dried under nitrogen flow. Samples were imaged in air in non-contact acoustic AC mode using Atomic force microscope 5500(Agilent Technologies). AC cantilever (normal spring constant of cantilever) was used. The topographic imaged of all samples were used for analysis using SPIP software.
Fluorescence Measurement
The binding of the compounds to α-synuclein was monitored by enhancement of drug fluorescence in the presence of equimolar quantity of protein. The samples were excited at the absorbance maxima of the respective compounds and the fluorescence spectra were recorded using using a Jobin Vyon Horiba Fluoromax-4 spectrofluorimeter.
Intrinsic Fluorescence
For monitoring the intrinsic fluorescence of α-synuclein, 200 μL of samples were prepared by incubating 70 µM protein with equal concentration of a given compound. Samples were excited at 280 nm, and spectra were recorded between 285 nm and 500 nm. All the experiments were performed at least three times.
Dot-Blot Assay
α-synuclein samples (3 μL) were spotted onto a nitrocellulose membrane and were allowed to dry at room temperature. The nitrocellulose membrane was incubated in 10% BSA dissolved in 0.1% Tween 20, Tris-buffered saline (TBS-T) solution for 1 h. The blocking solution was removed and the membrane was washed three times for 5 min each with TBS-T solution. The membrane was then incubated in antibody solution for 1 h. The A11 antibody was diluted in 5% TBS-T solution according to the manufacturer’s recommendation. After incubation, the membrane was washed three times for 5 min using TBS-T solution. The membrane was incubated in (1:10,000 dilution in 5% BSA TBS-T) HRP-conjugated IgG for 1 h. Then the membrane was washed three times for 5 min each with PBS-T solution and the same detection method as previously described was used. The blot images were captured using a Digital imaging system –ImageQuant™ LAS 4000.
Cell Culture
The human neuroblastoma cell line SH-SY5Y was obtained from American Type Culture Collection (ATCC). Cells were grown at 37°C in a humidified incubator with 5% CO2 in Dulbecco’s modified Eagle medium (DMEM, GIBCO) supplemented with 10% fetal bovine serum (FBS, GIBCO) and 1 mM glutamine.
Determination of Cytotoxicity
Cytotoxicity was evaluated with the colorimetric MTT [3-(4, 5-dimethyl-2-thiazolyl)- 2, 5-diphenyl-2Htetrazolium bromide] assay. SH-SY5Y cells were seeded at 5 × 104 cells/well in 96-well plates. After 12 hr, the medium was removed and replaced with media containing protein alone or with the tested compounds and left at 37°C. After 24 hr, spent medium was replaced with 90 μL of fresh medium and 10 μL of MTT (5 mg/mL) in PBS (0.5 mg/mL, final concentration). The cells were incubated for another 4 hr. After the removal of MTT, formazan crystals formed were dissolved in acidified isopropanol. The amount of formazan was evaluated by measuring the optical density at 570 nm. Cell viability was expressed as the percentage of control cells and was calculated using the equation (5)

Where C is cell viability, A is the absorbance of the treated neurons after subtracting the absorbance of only MTT and At is the absorbance of the untreated neurons after subtracting the absorbance of only MTT.
Proteinase-K Digestion
A 20 mg/mL stock of Proteinase-K was diluted to 50 µg/ mL and added to 60 µg of 30 days aggregated protein samples. Reaction was stopped by adding SDS-sample buffer after 1 h incubation at 37°C. Digested samples were run on a 12% SDS-PAGE and visualised with coomassie brilliant blue.
Turbidity Assay
5 μL of protein incubations were added to 145 μL of same buffer used for aggregation and absorbance at 360 nm was recorded as a measure of turbidity. Change in turbidity was calculated by subtracting the turbidity of the respective sample at the beginning of the experiment.
Statistical Analyses
Results were expressed as the means and the standard deviation (S.D.) values, with n as the number of experiments. Calculation of IC50 values from dose-dependency curves was performed using Sigma Plot 10.0. P values for significance were determined by unpaired Student’s t test. In all analyses, the null hypothesis was rejected at the 0.05 level.
Additional Information
How to cite this article: Ahsan, N.,Mishra, S., Jain,M.K., Surolia, A. & Gupta, S.Curcumin Pyrazole and its derivative (N-(3-Nitrophenylpyrazole) Curcumin inhibit aggregation, disrupt fibrils andmodulate toxicity ofWild type and Mutant α-Synuclein. Sci. Rep. 5, 9862; doi: 10.1038/srep09862 (2015).
References
Moreno-Gonzalez, I. & Soto, C. Misfolded protein aggregates: mechanisms, structures and potential for disease transmission. Semin Cell Dev Biol 22, 482–7 (2011).
Stefanova, N., Bucke, P., Duerr, S. & Wenning, G. K. Multiple system atrophy: an update. Lancet Neurol 8, 1172–8 (2009).
Jellinger, K. A. Neuropathological spectrum of synucleinopathies. Mov Disord 18 Suppl 6, S2–12 (2003).
George, S., Rey, N. L., Reichenbach, N., Steiner, J. A. & Brundin, P. alpha-Synuclein: the long distance runner. Brain Pathol 23, 350–7 (2013).
Marques, O. & Outeiro, T. F. Alpha-synuclein: from secretion to dysfunction and death. Cell Death Dis 3, e350 (2012).
Bayer, T. A. et al. Alpha-synuclein accumulates in Lewy bodies in Parkinson's disease and dementia with Lewy bodies but not in Alzheimer's disease beta-amyloid plaque cores. Neurosci Lett 266, 213–6 (1999).
Uversky, V. N. & Eliezer, D. Biophysics of Parkinson's disease: structure and aggregation of alpha-synuclein. Curr Protein Pept Sci 10, 483–99 (2009).
Volles, M. J. et al. Vesicle permeabilization by protofibrillar alpha-synuclein: implications for the pathogenesis and treatment of Parkinson's disease. Biochemistry 40, 7812–9 (2001).
Pieri, L., Madiona, K., Bousset, L. & Melki, R. Fibrillar alpha-synuclein and huntingtin exon 1 assemblies are toxic to the cells. Biophys J 102, 2894–905 (2012).
Lashuel, H. A. et al. Alpha-synuclein, especially the Parkinson's disease-associated mutants, forms pore-like annular and tubular protofibrils. J Mol Biol 322, 1089–102 (2002).
Li, J., Zhu, M., Rajamani, S., Uversky, V. N. & Fink, A. L. Rifampicin inhibits alpha-synuclein fibrillation and disaggregates fibrils. Chem Biol 11, 1513–21 (2004).
Cheng, B. et al. Inhibiting toxic aggregation of amyloidogenic proteins: a therapeutic strategy for protein misfolding diseases. Biochim Biophys Acta 1830, 4860–71 (2013).
Rigacci, S. et al. Abeta(1–42) aggregates into non-toxic amyloid assemblies in the presence of the natural polyphenol oleuropein aglycon. Curr Alzheimer Res 8, 841–52 (2011).
Garcia-Alloza, M., Borrelli, L. A., Rozkalne, A., Hyman, B. T. & Bacskai, B. J. Curcumin labels amyloid pathology in vivo, disrupts existing plaques and partially restores distorted neurites in an Alzheimer mouse model. J Neurochem 102, 1095–104 (2007).
Ringman, J. M., Frautschy, S. A., Cole, G. M., Masterman, D. L. & Cummings, J. L. A potential role of the curry spice curcumin in Alzheimer's disease. Curr Alzheimer Res 2, 131–6 (2005).
Qiu, X. et al. Synthesis and identification of new 4-arylidene curcumin analogues as potential anticancer agents targeting nuclear factor-kappaB signaling pathway. J Med Chem 53, 8260–73 (2010).
Ferrari, E. et al. Newly synthesized curcumin derivatives: crosstalk between chemico-physical properties and biological activity. J Med Chem 54, 8066–77 (2011).
Zhao, C., Liu, Z. & Liang, G. Promising curcumin-based drug design: mono-carbonyl analogues of curcumin (MACs). Curr Pharm Des 19, 2114–35 (2013).
Chakraborti, S. et al. Stable and potent analogues derived from the modification of the dicarbonyl moiety of curcumin. Biochemistry 52, 7449–60 (2013).
Mishra, S., Karmodiya, K., Surolia, N. & Surolia, A. Synthesis and exploration of novel curcumin analogues as anti-malarial agents. Bioorg Med Chem 16, 2894–902 (2008).
Selvam, C., Jachak, S. M., Thilagavathi, R. & Chakraborti, A. K. Design, synthesis, biological evaluation and molecular docking of curcumin analogues as antioxidant, cyclooxygenase inhibitory and anti-inflammatory agents. Bioorg Med Chem Lett 15, 1793–7 (2005).
Narlawar, R. et al. Curcumin-derived pyrazoles and isoxazoles: Swiss army knives or blunt tools for Alzheimer's disease? ChemMedChem 3, 165–72 (2008).
Changtam, C., Hongmanee, P. & Suksamrarn, A. Isoxazole analogs of curcuminoids with highly potent multidrug-resistant antimycobacterial activity. Eur J Med Chem 45, 4446–57 (2010).
Ono, K., Hasegawa, K., Naiki, H. & Yamada, M. Curcumin has potent anti-amyloidogenic effects for Alzheimer's beta-amyloid fibrils in vitro. J Neurosci Res 75, 742–50 (2004).
Hudson, S. A., Ecroyd, H., Kee, T. W. & Carver, J. A. The thioflavin T fluorescence assay for amyloid fibril detection can be biased by the presence of exogenous compounds. FEBS J 276, 5960–72 (2009).
Klunk, W. E., Jacob, R. F. & Mason, R. P. Quantifying amyloid by congo red spectral shift assay. Methods Enzymol 309, 285–305 (1999).
Caruana, M. et al. Inhibition and disaggregation of alpha-synuclein oligomers by natural polyphenolic compounds. FEBS Lett 585, 1113–20 (2011).
Chakraborti, S. et al. Curcumin recognizes a unique binding site of tubulin. J Med Chem 54, 6183–96 (2011).
Libertini, L. J. & Small, E. W. The intrinsic tyrosine fluorescence of histone H1. Steady state and fluorescence decay studies reveal heterogeneous emission. Biophys J 47, 765–72 (1985).
Zhu, M., Li, J. & Fink, A. L. The association of alpha-synuclein with membranes affects bilayer structure, stability and fibril formation. J Biol Chem 278, 40186–97 (2003).
Porat, Y., Abramowitz, A. & Gazit, E. Inhibition of amyloid fibril formation by polyphenols: structural similarity and aromatic interactions as a common inhibition mechanism. Chem Biol Drug Des 67, 27–37 (2006).
Nakagami, Y. et al. A novel beta-sheet breaker, RS-0406, reverses amyloid beta-induced cytotoxicity and impairment of long-term potentiation in vitro. Br J Pharmacol 137, 676–82 (2002).
Yang, F. et al. Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques and reduces amyloid in vivo. J Biol Chem 280, 5892–901 (2005).
Singh, P. K. et al. Curcumin modulates alpha-synuclein aggregation and toxicity. ACS Chem Neurosci 4, 393–407 (2013).
Cremades, N. et al. Direct observation of the interconversion of normal and toxic forms of alpha-synuclein. Cell 149, 1048–59 (2012).
Tsika, E. et al. Distinct region-specific alpha-synuclein oligomers in A53T transgenic mice: implications for neurodegeneration. J Neurosci 30, 3409–18 (2010).
Kayed, R. et al. Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol Neurodegener 2, 18 (2007).
Narhi, L. et al. Both familial Parkinson's disease mutations accelerate alpha-synuclein aggregation. J Biol Chem 274, 9843–6 (1999).
Wise-Scira, O., Dunn, A., Aloglu, A. K., Sakallioglu, I. T. & Coskuner, O. Structures of the E46K mutant-type alpha-synuclein protein and impact of E46K mutation on the structures of the wild-type alpha-synuclein protein. ACS Chem Neurosci 4, 498–508 (2013).
Recchia, A. et al. Alpha-synuclein and Parkinson's disease. FASEB J 18, 617–26 (2004).
Ahmad, B. & Lapidus, L. J. Curcumin prevents aggregation in alpha-synuclein by increasing reconfiguration rate. J Biol Chem 287, 9193–9 (2012).
Smith, W. W. et al. Endoplasmic reticulum stress and mitochondrial cell death pathways mediate A53T mutant alpha-synuclein-induced toxicity. Hum Mol Genet 14, 3801–11 (2005).
Acknowledgements
The present work is supported by NII Core and DBT grant (BT/PR12648/BRB/10/711/2009) to SG. This work is supported by an extramural grant for Bhatnagar Fellowship to AS by the Council of Scientific and Industrial Research (CSIR), India and from the core grant of NII. NA is a SRF of DBT, SM is a Senior Research Associate in the above grant of CSIR to AS. Authors wish to thank Professor Rajiv Bhat, School of Biotechnology, Jawaharlal University, New Delhi for the gift of A53T mutant derived from the Wild type alpha synuclein by MKJ and Rajiv Bhat. Wild type alpha synuclein was a gift from Professor Peter Lansbury, Department of Neurology, Harvard Medical School, USA. Authors thank Mrs. Rekha Rani for technical assistance in acquiring TEM images.
Author information
Authors and Affiliations
Contributions
AS and SG provided the entire infrastructure and supervised the research. NA and SG performed and analysed the experiments. SM synthesized the compounds. MKJ prepared the A53T mutant α-synuclein construct. NA, SG and SM wrote the manuscript. NA, SM, AS and SG reviewed the manuscript.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Ahsan, N., Mishra, S., Jain, M. et al. Curcumin Pyrazole and its derivative (N-(3-Nitrophenylpyrazole) Curcumin inhibit aggregation, disrupt fibrils and modulate toxicity of Wild type and Mutant α-Synuclein. Sci Rep 5, 9862 (2015). https://doi.org/10.1038/srep09862
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep09862
This article is cited by
-
Improved bioactivities of curcumin pyrazole and its HP\(\beta\)CD inclusion complex compared to curcumin
Journal of Chemical Sciences (2023)
-
The effects of nano-curcumin supplementation on adipokines levels in obese and overweight patients with migraine: a double blind clinical trial study
BMC Research Notes (2022)
-
Disease-modifying treatment of Parkinson’s disease by phytochemicals: targeting multiple pathogenic factors
Journal of Neural Transmission (2022)
-
Pharmacophore modeling and 3D-QSAR study for the design of novel α-synuclein aggregation inhibitors
Journal of Molecular Modeling (2021)
-
The inhibitory potency of isoxazole-curcumin analogue for the management of breast cancer: A comparative in vitro and molecular modeling investigation
Chemical Papers (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
