
Abstract
Thalassospiramides comprise a large family of lipopeptide natural products produced by Thalassospira and Tistrella marine bacteria. Here we provide further evidence of their nanomolar inhibitory activity against the human calpain 1 protease. Analysis of structure-activity relationship data supported our hypothesis that the rigid 12-membered ring containing an α,β-unsaturated carbonyl moiety is the pharmacologically active functional group, in contrast to classic electrophilic “warheads” in known calpain inhibitors. Using a combination of chemical modifications, mass spectrometric techniques, site-directed mutagenesis and molecular modeling, we show the covalent binding of thalassospiramide's α,β-unsaturated carbonyl moiety to the thiol group of calpain's catalytic Cys115 residue by a Michael 1,4-addition reaction. As nanomolar calpain inhibitors with promising selectivity and low toxicity from natural sources are rare, we consider thalassospiramides as promising drug leads.
Similar content being viewed by others

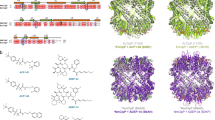
Introduction
Calpain is a calcium-dependent cysteine protease that participates in many signal transduction events by catalyzing the proteolysis of specific peptides in target substrates1,2,3. Deregulation of calpain action has been identified in pathologies such as neurological disorders, muscular dystrophies, cortical cataracts, cancer and inflammation4,5,6,7,8,9. To date, more than 200 calpain inhibitors have been reported with most being synthetic peptides and peptidomimetics that target active site residues10. A common feature of these inhibitors is the presence of a classical electrophilic warhead (e.g., aldehyde, α-ketocarbonyl and epoxysuccinyl) to interact with the active site cysteine residue (Cys115) of calpain11,12,13. However, major hindrances in the clinical application of these classical inhibitors are their poor selectivity for calpain, tendency to interact with other cysteine proteases and high potential for toxicity14,15,16.
Recently, we characterized 14 new and 2 known thalassospiramide lipopeptides from several Thalassospira and Tistrella marine bacterial species (see Fig. 1) and revealed their novel biosynthetic pathways17. Among these analogues, six were evaluated for their potent inhibitory activity against human calpain 1 protease (HCAN1). Although differences in bioactivity were as large as 20-fold, all tested thalassospiramides were active at nanomolar concentrations, which suggests they are thus far the most potent calpain inhibitors retrieved from natural sources13,14. Interestingly, the lack of the classical warhead and the presence of a common 12-membered ring system suggest that thalassospiramides may represent a new class of calpain inhibitors.

Chemical structure of thalassospiramide analogues.
All thalassospiramides share a rigid 12-membered ring and a variable lipopeptide side chain (R). See Ross et al.17 for full structures.
Results
Bioassay and Chemical Modifications
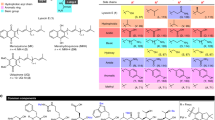
We accumulated all previously reported thalassospiramide analogues and evaluated their calpain 1 inhibitory activity using a fluorescence-based assay. The result showed that all thalassospiramides possessed nanomolar-level inhibitory activity against human calpain 1 (see Table 1), which suggests that the conserved 12-membered ring system with its electrophilic, unsaturated amide group might be the pharmacologically active moiety. To test this hypothesis, thalassospiramide A (1) was hydrolyzed at the ester position to 2 as well as hydrogenated at the double bond to 3 (see Fig. 2). In both cases, the products were 100-fold less active in the calpain inhibitory assay, strongly indicating that the intact 12-membered ring system is a critical element for the inhibitory activity. Reduction of 1 to 3 also resulted in the saturation of the acyl side chain, which, based on natural thalassospiramide analogues in the series, does not significantly influence the overall calpain bioactivity (see Table 1). These results supported our hypothesis that the α,β-unsaturated carbonyl moiety in the 12-membered ring system is essential for the inhibitory activity of calpain. We therefore predicted that Cys115 of calpain attacks the double bond of the unsaturated amide via a Michael-type 1,4-addition to form a covalent linkage between the inhibitor and protein. A similar binding mechanism was reported between the active site Thr1 residue of the 20S proteasome and the bacterial natural product syringolin A, a potent proteasome inhibitor that also contains an α,β-unsaturated amide in a 12-membered ring system18.

Chemical modifications of 1 and the comparison of IC50 values against HCAN1.
Both modifications (ester hydrolysis to 2 and double-bond saturation to 3) led to loss of bioactivity.
Human Calpain 1 Sample Analysis by MALDI-TOF MS
Top-down and bottom-up mass spectrometry analyses were next used to explore this postulated mode of action19,20,21,22. Excess 1 and 3 were added to HCAN1 and incubated separately before being subjected to MALDI-TOF analysis. The results revealed that 1 (957.5 Da) formed a covalent adduct with calpain, as the m/z value was shifted by approximately 974 Da in comparison to the control sample of “free” HCAN1 (see Fig. 3A). We measured only a 1:1 (HCAN1 to 1) complex despite using excessive amounts of 1, suggesting a specific interaction between HCAN1 and 1. Conversely, the HCAN1 + 3 complex did not yield a significant mass shift (see Fig. 3A), as anticipated, which is consistent with the loss of the electrophilic olefin in the 12-membered ring of 1. These findings support the specific binding of 1 to just a single calpain amino acid residue.

MALDI-TOF MS analysis of calpain samples.
A) Intact protein analysis of free HCAN1, HCAN1 + 3 and HCAN1 + 1 samples. Compared with free HCAN1, an obvious shift could be detected in the HCAN1 + 1 sample, which has a molecular weight similar to 1 (957.5 Da). B) Analysis of the three samples after trypsin digestion. Signal of original peptide (m/z = 3097.5) completely disappeared in the HCAN1 + 1 sample.
To explore the nature of the covalent linkage between thalassospiramide and calpain, we digested three samples (free HCAN1, HCAN1 + 1 and HCAN1 + 3) with trypsin and analyzed the products by MALDI-TOF/TOF MS. The results revealed that the Cys115-containing peptide fragment TDICQGALGDC115WLLAAIASLTLNDTLLHR (cal. m/z = 3097.6) could be detected in the free HCAN1 and HCAN1 + 3 samples but not in the HCAN1 + 1 sample (see Fig. 3B and Supplementary Fig. 1) in support of the proposed mechanism. Unfortunately, using MALDI-TOF we were unable to detect the anticipated tryptic peptide fragment with 1 (m/z = 4055.1)23,24.
Protein Mutation, Expression and Identification
Our previous MALDI-TOF MS result showed that only one molecule of 1 binds per calpain (see Fig. 3A), which indicates that the interaction between calpain and thalassospiramide is highly specific. If Cys115 is the specific attachment site, then a Cys115-mutant calpain should not form a covalent bond with 1 and therefore no mass shift should be observed. To test this hypothesis, the protease core of human calpain 1 (domains I-II of μ-calpain, “μI-II” as abbreviation), which retains the functional and structural elements of intact calpain25, was mutated to replace the Cys115 active site residue with Ala (μI-IIC115A). Native and mutated μI-II proteins were expressed, purified and their sequences verified (see Supplementary Fig. 3, 4 and 5). However, the calculated and observed masses of the two proteins, as measured by FT-ICR-MS, were in discordance (μI-II, cal. exact mass: 42239.9 Da, obs. exact mass: 42467.9 Da; μI-IIC115A, cal. Exact mass: 42207.9 Da, obs. exact mass: 42360.1 Da). Considering that the observed mass deviations of μI-II and μI-IIC115A were 228 and 152 amu, respectively and that both are multiples of 76, we reasoned that these differences were most likely due to the addition of β-mercaptoethanol (β-ME, 76.0 Da) from the storage buffer26. In order to verify that the observed mass deviations of the intact protein were actually caused by β-ME adducts, we performed a brief (30 minute) trypsin digestion of μI-II, to be followed by MS analysis, without the use of additional reducing agents such as TCEP27. HPLC fractionation and MS/MS analysis of the resulting peptides showed that three of the five cysteine residues were modified by β-ME (cys49, cys108 and Cys115; see Supplementary Fig. 6). This is consistent with the crystal structure of μI-II28 (PDB file: 1ZCM) in which only these three cysteine residues reside on the solvent-accessible surface of the protein. Verification of the formation of these β-ME adducts clearly explained the observed masses for both μI-II (+228 Da, 3x β-ME) and μI-IIC115A (+152 Da, 2x β-ME).
Expressed Protein Sample Analysis by FT-ICR and Q-TOF MS
We next incubated excess 1 with μI-II and μI-IIC115A, purified the samples by C4 RP-HPLC and analyzed them by FT-ICR-MS. HPLC chromatograms showed that the retention time of μI-II was changed by 0.3 min following the addition of 1 and a shift of +881.5 amu was clearly measured by MS (see Fig. 4 and Fig. 5). This mass shift represents the displacement of β-ME (loss of 76 Da) from Cys115 and the subsequent addition of 1 (addition of 957.5 Da) in its place. On the contrary, neither a change of HPLC retention time nor a mass shift in the MS signal was found for the μI-IIC115A sample after the addition of 1 (see Fig. 4 and Fig. 5). These results clearly signify that 1 interacts specifically with the Cys115 residue. As additional confirmation of these results, both μI-II + 1 and μI-IIC115A + 1 samples were digested overnight with trypsin and analyzed by LC-MS/MS (qTOF), which allowed the detection of the 1-modified fragment in the μI-II + 1 sample (see Fig. 6 and Supplementary Fig. 8) and not the μI-IIC115A + 1 sample. Based on these complimentary results, we conclude that thalassospiramide is exclusively associated with the Cys115 residue of calpain by a 1, 4-addition reaction.

HPLC comparison of μI-II and μI-IIC115A samples after the addition of 1.
The retention time of μI-II was shifted by 0.3 min, while no obvious shift can be observed in μI-IIC115A sample.

FT-ICR-MS analysis of μI-II, μI-II + 1, μI-IIC115A and μI-IIC115A + 1.
After the addition of 1, a 881.5 shift was clearly observed in μI-II samples (peak a: 42467.9; peak d: 43349.4), while there was no shift in μI-IIC115A samples (peak e: 42360.1 Da; peak g: 42360.1 Da). Mass spectra shown are deconvoluted with neutral monoisotopic masses displayed above each peak.

MS/MS annotation of 1-modified fragment in the μI-II + 1 sample (b ions of 1 labeled by b′ and red color).
Molecular Modeling Study
To gain further insight into the binding properties between thalassospiramide analogues and calpain, we performed large-scale docking between various thalassospiramides and 30 representative calpain conformations (PDB file: 1ZCM) generated via molecular dynamics simulations. Our docking results showed that thalassospiramide C (4), which experimentally demonstrated the greatest potency, reached Cys115 over the shortest distance, had the lowest docking energy of −8.1 kcal/mol and experienced the largest number of binding conformations with a distance of less than 5 Å (see Supplementary Table 1 and Supplementary Fig. 9). Moreover, 4 had the shortest lipopeptide side chain that enables tight interaction with the three important regions of calpain (S1, S2 and S3)14, which is consistent with our previous observations of markedly different bioactivities associated with the variable hydrophobic tails in the thalassospiramide series. In the case of syringolin A, which shares a similar ring structure to 4, Clerc et al. found that its proteasome inhibitory activity was enhanced more than 100-fold by modifications to the side chain29. Furthermore, the nucleophilic attack of Cys115 leads to the S-configuration of the resulting chiral center, the same as the study of Cysteine Cathepsins30.
Preliminary Selectivity and Toxicity Studies
Thalassospiramides 1 and 4 were inactive against the human 20S proteasome at 10 μM and have been reported to have weak activity against papain and none against trypsin31,32. In a preliminary toxicity study, we did not observe any obvious growth inhibition against the bacterial strains Staphylococcus aureus, the fungal strain Candida albicans and HeLa cells with 10 μM 1 and 4–8. In addition, Oh et al. reported that at a concentration of 10 μM of either 1 or 9 or 20 μM of 10, no cytotoxicity was observed in a mouse model33. These preliminary findings suggest that thalassospiramide may possess both low toxicity and good selectivity.
Discussion
We report that thalassospiramide natural products are potent inhibitors of calpain and function differently from standard calpain inhibitors. Most inhibitors possess highly reactive aldehyde groups that bind covalently to the active site of calpain, causing disadvantages such as non-selectivity, instability and excessive metabolism34,35,36,37. Although other examples of α,β-unsaturated amides have been reported as electrophilic inhibitors of cysteine proteases38,39,40, this is the first report that an α,β-unsaturated carbonyl moiety in a rigid ring system functions as an electrophilic warhead at nanomolar scale inhibitory activity against calpain. Interestingly, our previous biosynthetic study revealed that despite the great variety of side chains amongst the 16 known thalassospiramide analogues, the 12-membered macrolide ring is perfectly conserved17. These observations suggest that this core structure may possess important physiological function(s) and could serve as a new scaffold for drug design.
Methods
Fluorescence-based Calpain Activity Assay
Calpain 1 activity was measured according to the protocol provided with the Calpain Activity Assay Kit (K240-100, BioVision, USA). Tested compounds were dissolved in DMSO and serially diluted in methanol to obtain various concentrations (600 nM, 125 nM, 25 nM, 5 nM, 1 nM). The calpain inhibition assays were performed in 96-well plates. Each assay was initiated by combining 85 μL extraction buffer and 0.1 U HCAN1 (BioVision, USA). Subsequently, 1 μL test compound of the desired concentration, 10 μL reaction buffer and 5 μL calpain substrate (Ac-LLY-AFC) was added to each well. The plate was incubated at 37°C for 1 h in the dark. The test samples were then recorded using a fluorometer equipped with an excitation wavelength of 400 nm and an emission wavelength of 505 nm. Samples without HCAN1 or test compound were defined as blank controls and samples without test compounds were defined as negative controls. The reported IC50 values are the average of triplicate determinations.
Preparation of Compounds
All thalassospiramides in this study were obtained from the marine bacteria Thalassospira sp. CNJ328 and Tistrella bauzanensis TIO732917. Compounds were purified using RP HPLC (Luna 5 μ C18 (2), 250 × 1.00 mm, 100 Å, Phenomenex, Torrance CA) and analyzed using UPLC-ESI-HRMS (1.7 μm C18 column, 0.25 mL/min, gradient from 5% to 95% CH3CN with 0.1% TFA, 25 min). The NMR spectra were obtained using a Varian Inova 500 MHz spectrometer. All samples were dissolved in MeOH-d4 and chemical shifts were reported in ppm relative to TMS. The ESI-HRMS analysis was conducted using a Bruker Daltonics micrOTOF instrument (Bruker Daltonics GmbH, Bremen, German). The procedures of chemical modifications of 1 were described: (a) Ester hydrolysis of 12-membered ring of 1. Compound 1 (1 mg) was dissolved in 3 mL water solution (0.01 N NaOH, 50% methanol) and stirred for 2 hour at room temperature. After the reaction, the sample was neutralized and the solution was evaporated in vacuo. The product 2 (0.6 mg) was dissolved in MeOH and purified using RP-HPLC with an isocratic method of 45% CH3CN in water. (b) Hydrogenation of double bond of 1. Compound 1 was dissolved in ethanol and stirred for 5 hour together with 10% Pd/C catalyst under atmospheric H2 at room temperature. The catalyst was blocked with 0.25-μm filter membrane and the solvent was removed in vacuo. The reaction product was confirmed by an increased molecular weight of 4 daltons and MS/MS annotation by LC/MS analysis.
MALDI-TOF Mass Spectrometry Analysis
For sample preparation, the C4 Zip Tip (Millipore) was used for desalting and purification by washing with 20% acetonitrile water solution (0.1% TFA). Protein samples were eluted with 60% acetonitrile water solution (0.1% TFA) and dried under a stream of nitrogen. Treated proteins were dissolved in a saturated sinapinic acid (SA) matrix solution and applied to the MALDI ground steel plate (Bruker Daltonics). After air drying, the samples were analyzed in linear mode using an ultrafleXtreme MALDI-TOF-MS (Bruker Daltonics, Germany).
Trypsin Digestion for Free HCAN1, HCAN1 + 1 and HCAN1 + 3 Samples
Trypsin (mass spectrometry grade, Promega) was added to the 3 samples for digestion to a final proteinase: protein ratio of 1:50 (w/w) and incubated at 37°C for 24 hours. Digested samples were then desalted and purified using the C18 Zip Tip (Millipore), dissolved in saturated SA matrix solution and analyzed in reflectron mode and linear mode by MALDI-TOF/TOF-MS.
Expression, Purification and Identification of μI-II and μI-IIC115A
The cDNAs were obtained from HeLa cells by reverse transcription (SuperScript® III Reverse Transcriptase, Invitrogen™) and human μI-II was amplified by PCR using pfu DNA polymerase (Tiangen, China). The active site Cys115 was mutated to Ala by overlap PCR and confirmed by sequencing. The two DNA sequences were inserted into plasmid pet28a using BamHI and SalI and the recombinant plasmids were sequenced and BLAST against human calpain 1 (see Supplementary Fig. 2).
μI-II and μI-IIC115A were expressed in E. coli BL21 and grown on LB medium containing 30 μg/mL kanamycin at 37°C with shaking (225 rpms). Protein expression was induced by the addition of 0.5 mM IPTG at a bacterial concentration of OD600 0.6, followed by bacterial cell growth for an additional 18 h at 16°C before harvesting. Protein purification was conducted using Ni2+-affinity columns according to the manufacturer's instructions. Eluted protein was concentrated using a 30 K molecular weight filter and exchanged into storage buffer (20 mM imidazole-HCl, 5 mM β-mercaptoethanol, 1 mM EDTA, 1 mM EGTA, 30% glycol, pH 7.6).
The two expressed proteins were identified by SDS-PAGE and MS/MS annotation of trypsin digests. The map of SDS-PAGE showed that the molecular weight of two expressed proteins were 35–48 kDa (see Supplementary Fig. 3), consistent with the predicted molecular weight. Tryptic peptides of μI-II and μI-IIC115A were analyzed by LC-HR-MS/MS using a Bruker Daltonics micrOTOF-QII. The tryptic peptide coverage of each protein, as determined by MS1 and verified by MS2 and the annotation of active site containing peptides are shown in Supplementary Fig. 4 and Fig. 5.
Intact Protein Sample Preparation and Analysis by FT-ICR-MS
In preparation for FT-ICR-MS analysis, the 4 proteins samples (μI-II, μI-II + 1, μI-IIC115A and μI-IIC115A + 1) were purified by off-line HPLC (Agilent Infinity 1200 equipped with a multiple wavelength detector). Stock protein samples were thawed and then mixed with a 1 mM CaCl2 solution. A portion of μI-II and a portion of μI-IIC115A were each mixed with a molar excess of 1 and these were allowed to incubate at room temperature for an additional 10 minutes prior to HPLC purification. Samples, each containing 10 μg of protein, were injected onto a C4 RP-HPLC column (Jupiter 5 μ C4, 150 mm × 4.60 mm, 300 Å, Phenomenex). These samples were loaded at 1 ml/min of 90% mobile phase A (H2O + 0.1% TFA) and 10% mobile phase B (ACN + 0.1% TFA), which was held for 5 minutes, then eluted by increasing to 90% mobile phase B over 20 minutes. Absorbance at 220 nm was monitored and fractions of the HPLC eluent containing the major peaks were collected, flash frozen and lyophilized to dryness. Immediately prior to FT-ICR-MS analysis, lyophilized fractions were dissolved in 100 μl of a solution containing a mixture of 49.5% H2O, 49.5% methanol and 1% formic acid (all LC/MS grade). Samples were introduced into an LTQ-FT hybrid mass spectrometer with a 6.4 T magnet (Thermo Electron, North America) using a TriVersa NanoMate (Advion BioSciences, Inc.). The NanoMate was utilized in direct-infusion mode with a spray pressure of 0.3 psig and an ESI voltage of 1.4 kV. The LTQ-FT ion optics were first tuned to m/z 816 using cytochrome C. The LTQ capillary temperature was maintained at 200°C. For the acquisition of high-resolution FT-ICR-MS spectra of the intact protein samples the following settings were used: 200000 resolution, m/z range of 700–1400, 3 micro-scans per scan, max injection time of 8000 ms and an FT automatic gain control (AGC) of 8 e5. In order to improve the clarity of the FT-ICR measurements of the intact proteins, an isolation window of 100 m/z units was centered upon m/z 1015 in order to capture multiple charge states of the protein within each of the four samples and a minimum of 1000 scans were acquired and averaged (see Supplementary Fig. 7). Deconvolution of intact protein FT-ICR mass spectra was performed by using Xtract (Thermo Electron, Bremen, Germany).
Limited Trypsin Digestion and FT-ICR-MS Analysis of μI-II
A sample of protein μI-II was briefly digested using trypsin (Trypsin Singles, proteomics grade, Sigma-Aldrich). A final trypsin:protein ratio of 1:20 (w/w) was mixed and incubated at 37°C for 30 minutes in 40 mM ammonium bicarbonate buffer. The reducing agents DTT and TCEP were intentionally excluding as these reducing agents have been shown to remove β-ME from the cysteines27. The limited trypsin digest of μI-II was fractionated by HPLC using the same column and gradient that was utilized for the intact protein analysis. The eluent was collected in 1 ml fractions (1 min/fraction), flash frozen and lyophilized. The collected fractions were dissolved in 30–60 μL of the 49.5% H2O, 49.5% methanol and 1% formic acid mixture and analyzed by FT-MS using similar instrument parameters as previously outlined, with the exceptions being that the FT-ICR resolution was lowered to 50000 and the range of m/z 200–2000 was recorded for all fractions. Peptides were identified on the basis of their intact mass and β-ME modified peptides were verified by MS/MS fragmentation (see Supplementary Fig. 6).
Bottom-up Analysis of Protein Samples by LC-HR-MS/MS
In preparation for bottom-up analysis of the 4 proteins samples (μI-II, μI-II + 1, μI-IIC115A and μI-IIC115A + 1) each one was reduced with DTT, alkylated and incubated with a trypsin: protein ration of 1:20 (w/w) at 37°C for 18 hours. Samples were subsequently quenched with an equal volume of 10% formic acid. For LC-HR-MS/MS analysis and Agilent Infinity 1290 UPLC connected to a Bruker Daltonics micrOTOF-QII qTOF mass spectrometer. The trypsin digested samples were separated using an RP-C18 column (Luna 3 μ C18 (2), 100 mm × 2.0 mm, 100 Å, Phenomenex). Samples were loaded at 0.25 ml/min of 95% mobile phase A (100% H2O + 0.1% formic acid) and 5% mobile phase B (ACN + 0.1% formic acid), which was held for 3 minutes, then mobile phase B was increased to 65% over 30 minutes and then B was increased to 100% over 2 minutes. The micrOTOF-QII was configured to have a nebulizer pressure of 1.6 bar, dry gas temperature of 200°C, dry gas flow-rate of 7 L/min. Mass spectra were acquired with an MS1 spectral rate of 2 Hz, an MS2 rate of 3 Hz and the collision RF was stepped from 200 Vpp to 600 Vpp (increments of 100 V) during each MS2 event in order to maximize observed fragment ions. A "lock-mass" internal calibrant (m/z 922.01; CAS 58943-98-9) was continuously introduced into the mass spectrometer in order to maintain proper calibration throughout the run. The MS/MS annotation of thaA-modified peptide is detailed in Supplementary Fig. 8.
Molecular Dynamics Simulations and Docking Study of Calpain and Thalassospiramides
The currently available structures for calpain-1 are 1ZCM and 2ARY, from which we selected the former because it is a bound structure in which the ligand is covalently linked to Cys11528. To prepare the MD simulations, we removed the ligand and determined the protonation states of all histidines using the PDB2PQR server41. The protein was then solvated in a water box with 13309 SPC waters42 and 9 sodium ions to neutralize the system. The GROMACS 4.5.5 simulation package43 with the Amber99sb force field44 were used in the MD simulations due to their speed. The system was minimized with a steepest descent algorithm followed by 200 ps MD simulations with a position restraint for heavy protein atoms, under NPT conditions with 1 bar of pressure and a temperature of 310 K using a V-rescale thermostat45. The cut-offs for both VDW and short-range electrostatic interactions were set at 10 Å and long-range electrostatic interactions were treated according to the Particle-Mesh Ewald method46. Water molecules were constrained using the SETTLE algorithm47 and all protein bonds were constrained by the LINCS algorithm48. Finally, we performed three independent 50-ns NPT simulations with the same initial conformation (i.e., the last frame of the position restraint simulation) and different initial velocities. The conformations were saved every 5 ps.
All of the 30,000 MD simulation frames were collected and divided into 30 clusters according to the C-alpha of two flexible loops (residue 69–82 and 251–261) using the k-center algorithm49. The average RMSD within each cluster was approximately 1.5 Å. We chose one representative conformation from each cluster for flexible docking using AutoDock Vina.
We used AutoDock Tools (ADT) to prepare the proteins and ligands prior to docking. The ADT merged nonpolar hydrogens of proteins and automatically detected each bond type of the ligands. Proteins and ligands were added with Gasteiger partial charges50. AutoDock Vina, which significantly improved the average accuracy of the binding mode prediction compared to AutoDock 4, was applied for the docking study. The docking grid box was centered near Cys115 with a volume of 24*26*34 (A3) and each docking had 48 (parameter: exhaustive) parallel runs. The sequence index of the selected flexible residues was: 72, 79, 109, 115, 254, 260, 261, 272 and 29851.
Fluorescence-based 20S Proteasome Inhibition Assay
Thalassospiramide 1 and 4 were serially diluted in methanol to obtain various concentrations and then incubated with 1 nM 20S proteasome in 96-well plates (Tris 25 mM, pH 7.5; SDS 0.03%; EDTA 0.5 mM, final reaction volume 40 μL) for 15 min at 37°C. The specific fluorogenic proteasome substrate Suc-LLVY-AMC (10 μL, 200 mM) was added and incubated for another 15 min at 37°C and then the fluorescence in each well was measured. The assay wells without inhibitor were conducted as positive control (related to 100% of enzyme activity)52.
Preliminary Toxicity Assays
The preliminary toxicity assays included antimicrobial and cytotoxic assays. In antimicrobial assay, LB (10 g of tryptone, 5 g of yeast extract, 10 g of NaCl, 1 L of dd H2O) and potato dextrose medium (200 g of potato, 20 g of dextrose, 1 L of dd H2O) were used to inoculate strain S. aureus ATCC 43300 and C. albicans ATCC 76615 (28°C, for 12 h), respectively. The test samples were prepared at 1 mM in DMSO and added to the broth (96-well plate) at 1% (V/V, final concentration 10 μM). Then, the microbes were incubated at 28°C overnight. Cell growth was recorded by measuring the optical density at 600 nm and Penicillin (for S. aureus) and Natamycin (for C. albicans) were used as positive controls. Human HeLa cells were used in cytotoxic assay. Cells (80 μL, 1 × 105/mL) were planted into 96-well plates for 12 h, then the test samples were dissolved in DMSO and added to the assay medium to inoculate for another 48 h. The cell viability was assayed by the MTT method.
References
Sorimachi, H., Hata, S. & Ono, Y. Expanding members and roles of the calpain superfamily and their genetically modified animals. Exp. Anim. Tokyo 59, 549–566 (2010).
Saez, M. E., Ramirez-Lorca, R., Moron, F. J. & Ruiz, A. The therapeutic potential of the calpain family: new aspects. Drug Discov. Today 11, 917–923 (2006).
Croall, D. E. & Ersfeld, K. The calpains: modular designs and functional diversity. Genome Biol. 8, 11 (2007).
Saatman, K. E., Creed, J. & Raghupathi, R. Calpain as a therapeutic target in raumatic brain injury. Neurotherapeutics 7, 31–42 (2010).
Zatz, M. & Starling, A. Mechanisms of disease: calpains and disease. New Engl. J. Med. 352, 2413–2423 (2005).
Leloup, L. & Wells, A. Calpains as potential anti-cancer targets. Expert Opin. Ther. Targets 15, 309–323 (2011).
Bevers, M. B. & Neumar, R. W. Mechanistic role of calpains in postischemic neurodegeneration. J. Cereb. Blood Flow Metab. 28, 655–673 (2008).
Zadran, S., Bi, X. N. & Baudry, M. Regulation of calpain-2 in neurons: implications for synaptic plasticity. Mol. Neurobiol. 42, 143–150 (2010).
Libertini, S. J. et al. Evidence for calpain-mediated androgen receptor cleavage as a mechanism for androgen independence. Cancer Res. 67, 9001–9005 (2007).
Pietsch, M., Chua, K. C. H. & Abell, A. D. Calpains: Attractive targets for the development of synthetic inhibitors. Curr. Top. Med. Chem. 10, 270–293 (2010).
Cuerrier, D., Moldoveanu, T., Inoue, J., Davies, P. L. & Campbell, R. L. Calpain inhibition by alpha-ketoamide and cyclic hemiacetal inhibitors revealed by X-ray crystallography. Biochemistry 45, 7446–7452 (2006).
Qian, J. et al. Cocrystal structures of primed side-extending alpha-ketoamide inhibitors reveal novel calpain-inhibitor aromatic interactions. J. Med. Chem. 51, 5264–5270 (2008).
Donkor, I. O. A survey of calpain inhibitors. Curr. Med. Chem. 7, 1171–1188 (2000).
Donkor, I. O. Calpain inhibitors: a survey of compounds reported in the patent and scientific literature. Expert Opin. Ther. Patents 21, 601–636 (2011).
Stuart, B. G. et al. Molecular modeling: A search for a calpain inhibitor as a new treatment for cataractogenesis. J. Med. Chem. 54, 7503–7522 (2011).
Schneck, J. L. et al. Chemical mechanism of a cysteine protease, cathepsin C, as revealed by integration of both steady-state and pre-steady-state solvent kinetic isotope effects. Biochemistry 47, 8697–8710 (2008).
Ross, A. C. et al. Biosynthetic multitasking facilitates thalassospiramide structural diversity in marine bacteria. J. Am. Chem. Soc. 135, 1155–1162 (2013).
Groll, M. et al. A plant pathogen virulence factor inhibits the eukaryotic proteasome by a novel mechanism. Nature 452, 755–U7 (2008).
Shiraki, T. et al. Alpha, beta-unsaturated ketone is a core moiety of natural ligands for covalent binding to peroxisome proliferator-activated receptor. J. Biol. Chem. 280, 14145–14153 (2005).
Reverdy, C. et al. Discovery of specific inhibitors of human USP7/HAUSP deubiquitinating enzyme. Chem. Biol. 19, 467–477 (2012).
Ge, Y., Rybakova, I. N., Xu, Q. G. & Moss, R. L. Top-down high-resolution mass spectrometry of cardiac myosin binding protein C revealed that truncation alters protein phosphorylation state. Proc. Natl. Acad. Sci. U.S.A. 106, 12658–12663 (2009).
Zhang, J. A., Dong, X. T., Hacker, T. A. & Ge, Y. Deciphering modifications in swine cardiac troponin I by top-down high-resolution tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 21, 940–948 (2010).
Paul, B., Das, D., Ellington, B. & Marsh, E. N. G. Probing the mechanism of cyanobacterial aldehyde decarbonylase using a cyclopropyl aldehyde. J. Am. Chem. Soc. 135, 5234–5237 (2013).
Chait, B. T. Mass spectrometry: Bottom-up or top-down? Science 314, 65–66 (2006).
Moldoveanu, T. et al. A Ca2+ switch aligns the active site of calpain. Cell 108, 649–660 (2002).
Orsatti, L., Pallaoro, M., Steinkuler, C., Orru, S. & Bonelli, F. Reactivity of the NS2/3((907–1206)) ASK(4) protein with beta-mercaptoethanol studied by electrospray ion trap mass spectrometry. Rapid Commun. Mass Spectrom. 16, 1919–1927 (2002).
Begg, G. E. & Speicher, D. W. Mass spectrometry detection and reduction of disulfide adducts between reducing agents and recombinant proteins with highly reactive cysteines. J. Biomol. Tech. 10, 17–20 (1999).
Li, Q. S., Hanzlik, R. P., Weaver, R. F. & Schonbrunn, E. Molecular mode of action of a covalently inhibiting peptidomimetic on the human calpain protease core. Biochemistry 45, 701–708 (2006).
Clerc, J. et al. Synthetic and structural studies on syringolin A and B reveal critical determinants of selectivity and potency of proteasome inhibition. Proc. Natl. Acad. Sci. U.S.A. 106, 6507–6512 (2009).
Mertens, M. D. et al. A coumarin- labeled vinyl sulfone as tripeptidomimetic activity- based probe for cysteine cathepsins. ChemBioChem 15, 955–959 (2014).
Kaneko, I. et al. inventors; Sankyo Co., Ltd. assignee. New compound B1371A or B1371B and their production. Japanese patent JP 06,298,796 (A). 1994 Oct 25.
Osumi, H. et al. inventors; Sankyo Co., Ltd. assignee. New compound B1371E or B1371F and their production. Japanese patent JP 08,081,431 (A). 1996 Jun 23.
Um, S. et al. Thalassospiramide G, a new gamma-amino-acid-bearing peptide from the marine bacterium Thalassospira sp. Mar. Drugs 11, 611–622 (2013).
Kang, D. H. et al. Identification of 3-Acetyl-2-aminoquinolin-4-one as a novel, nonpeptidic scaffold for specific calpain inhibitory activity. J. Med. Chem. 52, 3093–3097 (2009).
Cuerrier, D. et al. Development of calpain-specific inactivators by screening of positional scanning epoxide libraries. J. Biol. Chem. 282, 9600–9611 (2007).
Shirasaki, Y. et al. Exploration of orally available calpain inhibitors 2: Peptidyl hemiacetal derivatives. J. Med. Chem. 49, 3926–3932 (2006).
Donkor, I. O., Assefa, H. & Liu, J. Structural basis for the potent calpain inhibitory activity of peptidyl alpha-ketoacids. J. Med. Chem. 51, 4346–4350 (2008).
Ettari, R. et al. Novel peptidomimetics containing a vinyl ester moiety as highly potent and selective falcipain-2 Inhibitors. J. Med. Chem. 52, 2157–2160 (2009).
Kathman, S. G., Xu, Z. & Statsyuk, A. V. A Fragment-based method to discover irreversible covalent inhibitors of cysteine proteases. J. Med. Chem. 57, 4969–4974 (2014).
Nonoo, R. H., Armstrong, A. & Mann, D. J. Kinetic template-guided tethering of fragments. Chemmedchem 7, 2082–2086 (2012).
Dolinsky, T. J., Nielsen, J. E., McCammon, J. A. & Baker, N. A. PDB2PQR: An automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 32 (Web Server issue) W665–7 (2004).
Berendsen, H. J. C., Grigera, J. R. & Straatsma, T. P. The missing term in effective pair potentials. J. Phys. Chem.-Us 91, 6269–6271 (1987).
Hess, B., Kutzner, C., van der Spoel, D. & Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447 (2008).
Hornak, V. et al. Comparison of multiple amber force fields and development of improved protein backbone parameters. Proteins 65, 712–25 (2006).
Berendsen, H. J. C., Postma, J. P. M., Vangunsteren, W. F., Dinola, A. & Haak, J. R. Molecular-dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 (1984).
Cerutti, D. S., Duke, R. E., Darden, T. A., & Lybrand, T. P. Staggered mesh ewald: An extension of the smooth particle-mesh ewald method adding great versatility. J. Chem. Theory Comput. 5, 2322–2338 (2009).
Miyamoto, S. & Kollman, P. A. Settle - an analytical version of the shake and rattle algorithm for rigid water models. J. Comput. Chem. 13, 952–962 (1992).
Hess, B., Bekker, H., Berendsen, H. J. C., Fraaije, J. G. E. M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472 (1997).
Bowman, G. R., Huang, X. H. & Pande, V. S. Network models for molecular kinetics and their initial applications to human health. Cell Res. 20, 622–630 (2010).
Morris, G. M. et al. AutoDock 4 and AutoDock Tools 4: automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791 (2009).
Trott, O. & Olson, A. J. Software news and update AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 31, 455–461 (2010).
Trivella, D. B. B. et al. Enzyme inhibition by hydroamination: Design and mechanism of a hybrid carmaphycin-syringolin enone proteasome inhibitor. Chem. Biol 21, 782–791 (2014).
Acknowledgements
The authors thank Dr. Jianwei Sun for discussions regarding the structural chemistry, Dr. Peggy Pai, Miss Xiaofen Liu and Miss Joyce Wong for technical assistance with the MALDI-TOF-MS, Dr. Daniela B.B. Trivella for 20S proteasome inhibition assay and HKUST Biosciences Central Research Facility for providing MS services. The work was supported by grants from the China Ocean Mineral Resources Research and Development Association (DY125-15-T-02), the King Abdullah University of Science and Technology (SA-C0040/UK-C0016), the National Natural Science Foundation of China (21273188) and the NIH (GM97509).
Author information
Authors and Affiliations
Contributions
L.L., M.M., Z.C., X.H., P.D., Y.X., B.M. and P.Q. designed the experiments, L.L, M.M., S.G., W.Z., G.Z., L.L.(IU). executed the experiments, L.L., M.M., S.G. analyzed data and prepared the figures and L.L. wrote the manuscript with the input from all authors.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
supporting information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Lu, L., Meehan, M., Gu, S. et al. Mechanism of Action of Thalassospiramides, A New Class of Calpain Inhibitors. Sci Rep 5, 8783 (2015). https://doi.org/10.1038/srep08783
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep08783
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
