
Abstract
Stable and efficient knockdown of multiple gene targets is highly desirable for dissection of molecular pathways. Because it allows sequence-specific DNA binding, transcription activator-like effector (TALE) offers a new genetic perturbation technique that allows for gene-specific repression. Here, we constructed a multicolor lentiviral TALE-Kruppel-associated box (KRAB) expression vector platform that enables knockdown of multiple gene targets. This platform is fully compatible with the Golden Gate TALEN and TAL Effector Kit 2.0, a widely used and efficient method for TALE assembly. We showed that this multicolor TALE-KRAB vector system when combined together with bone marrow transplantation could quickly knock down c-kit and PU.1 genes in hematopoietic stem and progenitor cells of recipient mice. Furthermore, our data demonstrated that this platform simultaneously knocked down both c-Kit and PU.1 genes in the same primary cell populations. Together, our results suggest that this multicolor TALE-KRAB vector platform is a promising and versatile tool for knockdown of multiple gene targets and could greatly facilitate dissection of molecular pathways.
Similar content being viewed by others


Introduction
Hematopoietic stem cells (HSCs) are rare self-renewing and multipotential cells localized within the osteoblastic and vascular niches of adult bone marrow (BM)1,2,3. These cells can differentiate into all blood cell lineages and are therefore crucial for reconstitution of hematopoiesis into transplanted recipients with BM ablation4. HSCs are heterogeneous, consisting of long-term self-renewing HSCs (LT-HSCs) and short-term self-renewing HSCs (ST-HSCs). HSCs are rare, constituting between 0.05% to 0.1% of total murine BM cells and are quiescent under normal condition5,6; However, they can be enriched by cell sorting and reenter into the cell cycling in cell culture medium with addition of cytokines7. In vitro reactivated HSCs are amenable to retrovirus or lentivirus infection and are able to repopulate the whole BM when transplanted into lethally irradiated recipient mice. Thus, the ability of HSC in vitro activation and transplantation offers a convenient approach for gain- and loss-of-function studies in the hematopoietic compartment that includes HSCs.
Lentivirus- or retrovirus-mediated shRNA hairpins offer an effective approach for knockdown of gene targets in hematopoietic compartment, when combined with HSC activation and transplantation8. shRNA-based knockdown is dependent on Dicer and the RNA-induced silencing complex (RISC) to degrade mRNA targets. Therefore, alternative technologies that repress gene expression through different mechanisms from shRNA-based knockdown are also highly desirable in the field of biomedical research. Furthermore, it is worthwhile exploring the ability of these new technologies in knockdown of multiple gene targets.
Transcription activator-like effector (TALE) is a highly specific and high-affinity DNA-binding protein. When fused with a functional domain, it can regulate expression of endogenous genes or edits genome. TALE consists of tandem, highly conserved 33–34 amino repeats9,10. TALE can be engineered to bind virtually any desired DNA sequence and thus can regulate expression of specific endogenous gene when tethered with transcription activator (e.g. VP16, VP64) or repressor domains such as the Kruppel-associated box (KRAB) repressor domain11,12. Recently, we developed a vector for quick assembly of TALE-KRAB using the Golden Gate TALLEN2.0 cloning system and knocked down expression of the miR-302/367 cluster using TALE-KRAB targeting the promoter region of the miR-302/367 cluster12. Here, we developed a multicolor panel of lentiviral TALE-KRAB expression vectors for knockdown of multiple gene targets. Our data show the successful knockdown of the two gene targets (c-Kit and PU.1) in bone marrows of recipient mice, by combining TALE-KRAB based transcriptional repressors and bone marrow transplantation approach. We also demonstrate that this platform can simultaneously knock down both c-Kit and PU.1 genes in the same primary cell population. Collectively, our multicolor TALE-KRAB vector platform is a promising and versatile tool for knockdown of multiple gene targets and it will facilitate dissection of molecular pathways.
Results
Construction of a multicolor panel of lentiviral TALE-KRAB vectors for knockdown of multiple genes
TALE-based transcriptional repressor is a novel tool for gene knockdown11,12. We previously knocked down the miR-302/367 cluster expression by designing a specific TALE in pLenti-EF1α-KRAB (GG) vector12. To facilitate knockdown of multiple gene targets in cells, we constructed a multicolor panel of lentiviral TALE-KRAB vectors by replacing eGFP with the genes carrying Cerulean, mCherry-IRES-Blast, or Venus-IRES-Zeocin expression cassettes. Therefore, the resulting vectors express four fluorescent proteins: eGFP, Cerulean, mCherry and Venus (Fig. 1A). In addition, pLV_TALE-KRAB-mCherry-Blast and pLV_TALE-KRAB-Venus-Zeocin vectors were equipped with two drug resistant genes Blast and Zeocin. We confirmed these vectors by Sanger sequencing and showed that these vectors could express expected fluorescent proteins when transfected into 293T cells (Fig. 1B).

Construction of a multicolor panel of lentiviral vectors expressing TALE-KRAB.
(A) Maps of TALE-KRAB lentiviral vectors containing a multicolor panel of fluorescent proteins. TALE is fused to the N-terminus of the KRAB transcriptional repressor domain. Individual fluorescent protein gene is fused in-frame with KRAB via T2A sequence. Drug resistant genes Blasti and Zeo are at the downstream of mCherry and Venus fluorescent protein genes and separated by an IRES sequence. EF1a promoter drives TALE-KRAB, fluorescent protein genes and drug resistant genes. Blasti, resistance to Blasticidin S; Zeo, resistance to Zeocin. IRES, an internal ribosome entry site. NLS, nuclear localization signal. (B) Fluorescent images of 293T cells transfected with individual TALE-KRAB expression vectors. 293T cells were transfected with indicated vectors and fluorescent images were taken using an Olympus IX71 equipped with a color camera at 24 hrs after transfection.
Knockdown of the endogenous gene targets c-Kit and PU.1 in vitro by TALE-KRAB transcriptional repressors
To determine if TALE-KRAB can be used to knock down gene targets in hematopoietic compartment of mice, we selected two target genes: c-kit and PU.1. These two genes are expressed in hematopoietic compartment and play important role in regulation of hematopoiesis. c-Kit is a receptor tyrosine kinase (RTK) and mainly expressed in HSCs and multipotential progenitors13,14. PU.1 (also called SFPI1) is an Ets-family transcription factor and expressed in HSCs, as well as multipotential progenitors, which is crucial for the generation of all hematopoietic lineages. Dysregutlation of PU.1 leads to blockage of lineage development and/or leukemogenesis15,16. We designed two TALEs that recognize specific sequences within the proximal promoter region of mouse c-Kit and PU.1 and then fused each TALE with the Kruppel-associated box (KRAB) transcriptional repressor domain11,12,17 (Fig. 2A).

Knockdown of c-Kit and PU.1 by specific TALE-KRAB transcriptional repressors.
(A) Design of TALE-KRAB transcriptional repressors for knockdown of c-Kit and PU.1. A DNA-binding domain was designed to bind the indicated target sequence within the proximal promoter region upstream of the transcription start of mouse c-Kit and PU.1 genes, respectively. (B) Diagrams of luciferase reporters driven by promoters of mouse c-Kit and PU.1 genes. DNA fragment covering the proximal promoter region of c-Kit and PU.1 were amplified by PCR and cloned into pGL3-basic reporter vector and resultant reporters were designated as c-Kit-Luc and PU.1-Luc, respectively. The target sites of TALE1 and TALE2 for each gene were indicated. (C) Luciferase reporter assay of the c-Kit promoter-driven reporter (c-Kit-Luc). 293T cells were transfected with c-Kit-Luc and plasmids expressing control-TALE-KRAB, c-Kit-TALE1-KRAB, or c-Kit-TALE2-KRAB, respectively. pCMV-LacZ was included in each transfection as an internal control to normalize luciferase activity. Data represent the mean ± SD (n = 3, independent transfection replicates). ** p < 0.01, Student's t test. (D) Luciferase reporter assay of the PU.1 promoter-driven reporter (PU.1-Luc). 293T cells were transfected with PU.1-Luc and expression plasmids Control-TALE-KRAB or PU.1-TALE2-KRAB, respectively. pCMV-LacZ was included in each transfection as an internal control to normalize luciferase activity. Data represent the mean ± SD (n = 3, independent transfection replicates). ** p < 0.01, Student's t test. (E) qPCR analysis of c-Kit expression in primary BM cells expressing c-Kit specific TALE-KRAB. Fresh isolated primary BM cells were infected with TALE-Control-KRAB, c-Kit-TALE1-KRAB or c-Kit-TALE2-KRAB retroviruses. Transcripts of c-Kit were analyzed by qPCR. Data represent the mean ± SD (n = 3, technical replicates). ** p < 0.01, Student's t test. (F) qPCR analysis of PU.1 expression in primary BM cells expressing TALE-KRAB specific for PU.1 promoter. Fresh isolated primary BM cells were infected with retroviruses expressing Control-TALE-KRAB, PU.1-TALE1-KRAB or PU.1-TALE2-KRAB. Transcripts of PU.1 were analyzed by qPCR. Data represent the mean ± SD (n = 3, technical replicates). ** p < 0.01, Student's t test.
To examine inhibitory function of the designed TALE-KRAB constructs, we also generated two luciferase reporters for c-Kit and PU.1 promoters, respectively (Fig. 2B). Our luciferase reporter assay revealed that both c-Kit-TALE1-KRAB and c-Kit-TALE2-KRAB repressed the luciferase activity of the c-Kit reporter more than 20-fold, when compared to the control-TALE-KRAB group in 293T cells (Fig. 2C). Similarly, PU.1-TALE2-KRAB inhibited the activity of PU.1-Luc reporter by at least 16-fold (Fig. 2D). To further determine if both TALE-KRAB plasmids are capable of reducing expression of endogenous c-Kit and PU.1 genes, primary bone marrow cells were infected multiple times with retroviruses expressing control-TALE-KRAB, c-Kit-TALE1-KRAB, c-Kit-TALE2-KRAB, PU.1-TALE1-KRAB, or PU.1-TALE2-KRAB. Four days after last infection, we sorted GFP+ infected cells and measured the transcripts of c-Kit and PU.1 by quantitative PCR (qPCR). The results showed that both TALE-KRABs indeed reduced expression of c-Kit or PU.1 by more than 90%, when compared with the control-TALE-KRAB (Fig. 2E, F).
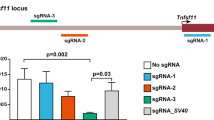
Recently, the RNA-guided CRISPR/Cas9 nuclease system was also modified as a genome-editing tool18,19,20. Different from TALEN, nuclease Cas9 protein is dependent on the engineered single guide RNA (gRNA) to bind with its target genomic DNA sequence. A nuclease-deficient Cas9 or dCas9 has been generated and this mutant Cas9 retains its DNA binding ability. When fused with effector domains such as VP64 and KRAB, dCas9-transcription factors can either active or repress expression of endogenous target genes21,22,23,24. Thus, we designed two guide RNAs (gRNAs) that target c-Kit promoter and then tested whether dCas9-KRAB could inhibit c-Kit promoter activity. Our luciferase reporter assay showed that RNA-guided dCas9-KRAB repressed activity of c-Kit promoter-driven luciferase reporter by two-folds (Fig. S1).
In vivo knockdown of the endogenous c-Kit and PU.1 genes by TALE-KRAB transcriptional repressors
Next, we assessed the ability of corresponding TALE-KRAB to reduce the expression of c-Kit and PU.1 in vivo. We prepared bone marrow mononuclear cells (BMNCs) from donor wild-type mice (CD45.2) seven days after 5-FU injection. We then stimulated BMNCs with three cytokines (IL-3, IL-6 and SCF) for twenty-four hours and infected them with retroviruses expressing TALE-KRAB specific for c-kit and PU.1 promoters. Infected cells were transplanted into lethally irradiated recipient mice (Fig. 3A). One month after BM transplantation, we sorted GFP-positive cells and analyzed expression of c-kit and PU.1 genes by qPCR analysis. Our results showed that the transcripts of c-kit and PU.1 were reduced 65% by corresponding TALE-KRAB, when compared with control-TALEKRAB (Fig. 3B, C). Because c-kit is a membrane-bound receptor, we measured c-kit expression on hematopoietic stem and progenitor cells from these recipient mice by flow cytometry analysis (Fig. 3D). Our data indicated that c-kit protein expression was decreased more than 4-fold by c-kit-TALE1-KRAB and c-kit-TALE2-KRAB on the membrane of progenitor cells (Lin−, left panel), stem and progenitor cells (Lin−Sca-1+, middle panel) and HSCs (Lin− CD150+CD48−, right panel). Taken together, our data demonstrated that TALE-KRAB mediated knockdown, combined with BM transplantation, could be used as a general approach for loss-of-function studies in hematopoietic compartment.

In vivo knockdown of c-Kit and PU.1 by TALE-KRAB transcriptional repressor.
(A) Diagram of BM transplantation assay. Donor BM cells that were transduced with specific c-Kit-TALE1/2-KRABs were injected together with helper BM cells into recipient mice that received a prior 9-Gy irradiation. Reconstituted mice were sacrificed for isolating BM cells at 1 month after BM transplantation and then performed immunostaining and flow cytometery analysis. The drawing was made by Z.Z. and W.S. using Adobe Illustrator and PowerPoint. (B) qPCR analysis of in vivo PU.1 expression in fresh BM cells expressing Control-TALE-KRAB, PU.1-TALE1-KRAB or PU.1-TALE2-KRAB. Reconstituted mice were sacrificed at 1 month after BM transplantation and then sorted GFP-positive-Lin- BM cells for qPCR. Data represent the mean ± SD (n = 3, technical replicates). ** p < 0.01, Student's t test. (C) qPCR analysis of in vivo c-Kit expression in primary BM cells expressing Control-TALE-KRAB, c-Kit-TALE1-KRAB or c-Kit-TALE2-KRAB. Reconstituted mice were sacrificed at 1 month after BM transplantation and then sorted GFP-positive-Lin- BM cells for qPCR. Data represent the mean ± SD (n = 3, technical replicates). ** p < 0.01, Student's t test. (D) Determination of in vivo c-Kit expression by flow cytometry. Total BM cells were prepared from reconstituted mice at 1 month after BM transplantation, which carried the Control-TALE-KRAB, c-Kit-TALE1-KRAB or c-Kit-TALE2-KRAB. The mean fluorescent intensity for c-Kit expression in various HSPC populations is displayed. Data represent the mean ± SD (n = 3). ** p < 0.01, Student's t test.
Simultaneous knockdown of PU.1 and c-Kit genes in primary cells
At last, we decided to test whether these multicolor TALE-KRAB vectors can enable simultaneous knockdown of two or more gene targets, such as PU.1 and c-Kit. To this end, we cloned PU.1-TALE2 and c-Kit-TALE2 into pLV_TALE-KRAB-mCherry-Blast and pLV_TALE-KRAB-Venus-Zeocin vectors, respectively (Fig. 4A). We then examined expression of PU.1 and c-Kit in HPSCs and mouse embryonic fibroblasts (MEFs). Our data showed that these two genes are expressed in both HPSCs and MEFs (Fig. 4A). We selected MEFs for our double knockdown experiment since these cells are relatively easy to be infected with viruses. We analyzed infection efficiency three days after coinfection with viruses expressing PU.1-TALE1-eGFP, PU.1-TALE2-mCherry and c-Kit-TALE2-Ceru (Fig. 4C). Our data showed that these three viruses achieved 68.2%, 30.8% and 36.9% infection efficiency, respectively. To sort MEFs co-expressing the three-fluorescence proteins, we first gated GFP-positive cells and then gated the cells positive for both Cerulean and mCherry in the GFP-positive cell population (Fig. 4D). By qPCR analysis, we analyzed the gene expression of c-Kit and PU.1 in the sorted MEFs. Our qPCR analysis showed that the expression of c-Kit and PU.1 genes were knocked down 50% and 80%, respectively, in the MEFs that were triple-positive for Green, Cerulean and mCherry fluorescence proteins.

Double knockdown of PU.1 and c-Kit genes in primary mouse embryonic fibroblasts (MEFs).
(A) RT-PCR analysis of c-Kit and PU.1 expression in primary cells. cDNA from MEFs and HSPCs were used for PCR amplification by c-Kit and PU.1 specific primers. HPRT was included as a control. The gels for each of the three genes (c-Kit, PU.1 and HPRT) were run under the same experimental conditions. (B) Diagram of PU.1-TALE1, 2 and c-Kit-TALE2. PU.1-TALE1 and PU.1-TALE2 were cloned into TALE-KRAB-eGFP and TALE-KRAB-mCherry-Blast, respectively. c-Kit-TALE2 was cloned into TALE-KRAB-Ceru. (C-D) Flow analysis and FACS of MEFs infected with viruses expressing TALE-KRAB repressors. MEFs were coinfected with three viruses expressing PU.1-TALE1-KRAB-eGFP, PU.1- TALE2-KRAB-mCherry-Blast and c-Kit-TALE2-KRAB-Ceru, respectively. Infected MEFs were analyzed by flow cytometer 7 days after infection (C). MEFs co-expressing c-Kit- and PU.1-TALE-KRAB transcriptional repressors were sorted by FACS (D). (E) qPCR analysis of c-Kit and PU.1 expression in MEFs co-expressing PU.1-TALE1, 2 and c-Kit-TALE2. Data represent the mean ± SD (n = 3). ** p < 0.01, Student's t test.
Discussion
TALEs are natural effector proteins originally secreted by Xanthomonas and Ralstonia bacteria, they can regulate gene expression in host plants and thus facilitate bacterial survival and colonization9,10. Significant efforts have been made toward genetic modification and transcriptional modulation by using TALE technology because designing TALE is much easier than zinc fingers and meganucleases12,25. We recently demonstrated that KRAB repressor domain fused with the TALE sequences designed specifically to target the promoter of miR-302/367 cluster is able to inhibit the expression level of endogenous miR-302/367 cluster in somatic cells12. In our current study, we combined BM transplantation with TALE-KRAB technology to knock down two targets c-kit and PU.1 in hematopoietic compartment in vivo. Our data showed that the expression of c-Kit and PU.1 were decreased more than 90% when compared with the control-TALE-KRAB group. It has been shown that TALE-VP64, which contains a TALE DNA binding domain fused to the VP64 transcription domain, is a powerful tool to selectively activate transcription of endogenous genes26. Thus, it is logical to redesign a new TALE-VP64 platform by replacing KRAB domain in our current TALE-KRAB vector system with VP64 activation domain, so that it will match with the Golden Gate TALEN and TAL Effector Kit 2.027.
We also showed that RNA-guided dCas9-KRAB could inhibit c-Kit promoter activity by a luciferase reporter assay. Our data indicate that TALE-KRAB is more effective than dCas9-KRAB in inhibiting c-Kit promoter activity. However, our data are not sufficient for making a general conclusion, because many factors such as target sites and sequences in promoter regions and loci of target genes can affect their inhibitory functions28.
Although siRNA/shRNA technology is a widely used approach for knockdown, it remains a technical challenge to silence genes with high specificity and efficiency, because specificity is inversely affected by the dosage of these inhibitors delivered into each cell29,30,31. In contrast, TALE-KRAB transcriptional repressors directly bind to the promoter region and inhibit mRNA expression of gene targets at transcriptional level. Thus, siRNA/shRNA and TALE-KRAB have two different classes of targets: mRNA and promoter DNA regions, respectively. It will be interesting to test whether the combination of TALE-KRAB and shRNA achieves a superior effect on knockdown of target genes in the future. Although we only knocked down two target genes in primary cells using our multicolor TALE-KRAB vector system in this current study, we believe that this system should facilitate knockdown of four target genes when assisted by FACS. In addition, the two TALE-KRAB vectors are equipped with Blasticidin and Zeocin resistant genes, which confer selection for cells stably expressing expected TALE-KRAB.
Recent studies show that human immunodeficiency virus type 1- (HIV-1) packaging and delivering of TALE nucleases (TALENs) into various human cell types can cause rearrangements of TALE repeats, whereas adenoviral vectors transfer intact TALEN genes32. Although we did not sequence the TALE repeats in host genome, our data indicate that delivering TALE repeats fused with KRAB and GFP into HSCs by lentivirus or retrovirus expression system can knock down the expression of target genes in vitro and in vivo. However, it remains to be tested if a packaging system is the primary cause for rearrangements of TALE repeats in cells.
In summary, we developed a multicolor TALE-KRAB expression vector platform that is compatible with the Golden Gate TALEN and TAL Effector Kit 2.027, an efficient and widely used method for TALE assembly. When combined with BM transplantation TALE-KRAB offers an effective and fast loss-of-function approach for investigating functional roles of multiple gene targets in hematopoietic compartment in vivo.
Methods
All the methods were carried out in “accordance” with the approved guidelines of the University of Illinois at Chicago.
Experimental animals
Mice were used for all the experiments when they were at 6 to 8 weeks of age. All mice studies were approved by the Animal Care and Use Committee at the University of Illinois at Chicago (approval number: 13–132).
Plasmid construction
pLenti-EF1α-KRAB-Ceru, pLenti-EF1α-KRAB-mCherry-Blasti and pLenti-EF1α-KRAB-venus-Zeo were constructed by replacing eGFP gene in pLenti-EF1α-KRAB (GG) vector with the following genes: cerulean fluorescent protein, mCherry-IRES-Blasti fusion and Venus-IRES-Zeo, respectively. A golden-gate cloning method was used to efficiently assemble TALEN constructs with custom repeat arrays27. Two TALE target sequences in the promoter regions of both c-Kit and PU.1 were selected using the TALE-NT program33. The TALE repeat arrays was assembled using the Golden Gate TALEN 2.0 kit (Addgene, Cambridge, MA) according to the published procedure27 and cloned into Esp3I sites of pLenti-EF1α-KRAB (GG) vector12. After confirming whole TALE repeats by sequencing, specific TALE-KRAB expression cassettes could be isolated by double digestion with BsiW I and EcoR I and cloned into pMIGR1 for retrovirus-mediated expression of TALE-KRAB.
To construct c-Kit and PU.1 reporter, ~1 kb promoter region of both target genes was amplified by PCR using specific primers (Table S1) and cloned into pGL3-basic vector (Promega). The resultant plasmids were designated as c-Kit-Luc and PU.1-Luc, respectively.
To construct c-Kit promoter-specific guide RNA (gRNA) expression plasmids, target sequences for gRNA1 and gRNA2 were selected by the online software (http://crispr.mit.edu) and cloned into pU6-gRNA vector. The resultant plasmids were named as c-Kit-gRNA1 and 2. CD48-gRNA1 was also constructed as a control. The expression plasmid dCas9-KRAB was constructed by replacing VP64 in pMLM3705 vector18 with a PCR product encoding KRAB domain12.
Cell culture
293T cells were maintained in DMEM (high glucose) containing 10% fetal bovine serum (FBS). Enriched fresh hematopoietic stem and progenitor cells were cultured in vitro in DMEM/F12 medium (7.5% FBS, 7.5% horse serum) plus 50 ng/ml SCF, 20 ng/ml IL-6 and 10 ng/ml IL-3. Culture medium was changed every other day. All cell culture products were purchased from Invitrogen except where noted otherwise.
Luciferase reporter transfection and luciferase assay
293T cells were cultured in 24-well plate overnight and then co-transfected with 100 ng of luciferase reporter, 350 ng of Control-TALE-KRAB or c-Kit-TALE1/2-KRAB or PU.1-TALE1/2-KRAB and 50 ng of pCMV-LacZ by using Fugene HD (Roche). Similarly, 293T were transfected with c-Kit-Luc reporter (100 ng), dCas9-KRAB expression plasmid (230 ng) and one of c-Kit-gRNA1, c-Kit-gRNA2 and CD48-gRNA1 expression plasmids or pU6-gRNA vector (120 ng). After 48 hours of transfection, cells were lysed in 250 μl of the passive lysis buffer (Promega) and assayed with a luciferase assay kit (Pormega) as directed by the manufacturer. Luciferase activities were expressed as relative luciferase/LacZ activities and normalized to those of control transfections in each experiment.
RNA extraction and RT-PCR
All the total RNAs were extracted using Trizol (Invitrogen) according to the manufacturer's instructions, with glycogen used as a carrier for precipitation. For Real-time qPCR analysis of target genes, 100 ng of total RNA was reverse-transcribed using Superscript III First-Strand Systhesis System (Invitrogen). The list of primers is included in Table S1.
Bone marrow transplantation
We used 8–12 week-old B6.SJL-Ptprca Pep3b/BoyJ (The Jackson Laboratory), a congenic C57BL/6 strain (CD45.1), as recipients for bone marrow (BM) transplantation experiments. For BM transplantation, recipient mice (4–8 weeeks old) were given acidified antibiotic water (1.1 g/L neomycin sulfate and 106 U/L polymyxin B sulfate) for 3 days and then irradiated with 9 Gy total body irradiation before BM transplantation by retro-orbital injection. Recipients were continuously fed with acidified antibiotic water for 30 days to reduce the chance of spontaneous infection.
Lentiviral and retroviral transduction of hematopoietic cells
Hematopoietic stem and progenitor cells were enriched from bone marrow of mice34 with the SpinSep™ Progenitors Enrichment Kit (StemCell Technologies). The enriched cells were cultured overnight and were spun at 200 × g. The supernatant was aspirated and replaced with the infection mixture consisting of 1 ml viral supernatant containing 5 μg/ml Ploybrene (Sigma), followed by centrifugation (900 g for 45 min). Two to four days post-infection, the cells were performed BM transplantation.
Analysis of hematopoietic stem and progenitor cells
BM were flushed from femurs of recipient mice with PBS containing 2% FBS, filtered through a nylon mesh and the red blood cells lysed. 2 × 106 cells were blocked with 5% rat serum and Fc-block for 10 min before antibody staining. For analysis of hematopoietic stem and progenitor cells (HPSCs), cells were stained for 20 min on ice with biotinylated antibodies specific for lineage markers (CD3e, B220, TER119, CD11b, Gr-1 and IL-7R), washed with PBS containing 2% FBS and then stained with two combinations (i.e. Streptavidin-V421, APC-anti-Sca-1, PE/Cy7-anti-c-Kit and 7-AAD, or Streptavidin-V421, APC-anti-CD150, PE-anti-CD48, PE/Cy7-anti-c-Kit and 7-AAD). The stained cells were analyzed with the Flow cytometer Fortessa LSR II instrument (Becton and Dickinson, CA). All of the antibodies were from Biolegend.
References
Sun, Y., Shao, L., Bai, H., Wang, Z. Z. & Wu, W. S. Slug deficiency enhances self-renewal of hematopoietic stem cells during hematopoietic regeneration. Blood 115, 1709–1717, 10.1182/blood-2009-07-232934 (2010).
Lo Celso, C. & Scadden, D. T. The haematopoietic stem cell niche at a glance. J. Cell Sci. 124, 3529–3535, 10.1242/jcs.074112 (2011).
Yin, T. & Li, L. The stem cell niches in bone. J. Clin. Invest. 116, 1195–1201, 10.1172/JCI28568 (2006).
Reya, T., Morrison, S. J., Clarke, M. F. & Weissman, I. L. Stem cells, cancer and cancer stem cells. Nature 414, 105–111, 10.1038/35102167 (2001).
Akashi, K., Traver, D., Miyamoto, T. & Weissman, I. L. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature 404, 193–197, 10.1038/35004599 (2000).
Kiel, M. J. et al. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 121, 1109–1121, 10.1016/j.cell.2005.05.026 (2005).
Yamazaki, S. et al. Cytokine signals modulated via lipid rafts mimic niche signals and induce hibernation in hematopoietic stem cells. EMBO J. 25, 3515–3523, 10.1038/sj.emboj.7601236 (2006).
Laurenti, E. et al. Inducible gene and shRNA expression in resident hematopoietic stem cells in vivo. Stem Cells 28, 1390–1398, 10.1002/stem.460 (2010).
Boch, J. et al. Breaking the code of DNA binding specificity of TAL-type III effectors. Science 326, 1509–1512, 10.1126/science.1178811 (2009).
Moscou, M. J. & Bogdanove, A. J. A simple cipher governs DNA recognition by TAL effectors. Science 326, 1501, 10.1126/science.1178817 (2009).
Cong, L., Zhou, R., Kuo, Y. C., Cunniff, M. & Zhang, F. Comprehensive interrogation of natural TALE DNA-binding modules and transcriptional repressor domains. Nature Commun. 3, 968, 10.1038/ncomms1962 (2012).
Zhonghui Zhang, D. X., Fransisca Heriyanto, Yongxing Gao, Zhijian Qian, Wen-Shu Wu . Dissecting the Roles of miR-302/367 Cluster in Cellular Reprogramming Using TALE-based Repressor and TALEN. Stem Cell Reports 1, 218–225 (2013).
Ikuta, K. & Weissman, I. L. Evidence that hematopoietic stem cells express mouse c-kit but do not depend on steel factor for their generation. P. Natl. Acad. Sci. USA. 89, 1502–1506 (1992).
Kimura, Y. et al. c-Kit-mediated functional positioning of stem cells to their niches is essential for maintenance and regeneration of adult hematopoiesis. PloS One 6, e26918, 10.1371/journal.pone.0026918 (2011).
Nutt, S. L., Metcalf, D., D'Amico, A., Polli, M. & Wu, L. Dynamic regulation of PU.1 expression in multipotent hematopoietic progenitors. J. Exp. Med. 201, 221–231, 10.1084/jem.20041535 (2005).
Rosenbauer, F. et al. Acute myeloid leukemia induced by graded reduction of a lineage-specific transcription factor, PU.1. Nat. Genet. 36, 624–630, 10.1038/ng1361 (2004).
Margolin, J. F. et al. Kruppel-associated boxes are potent transcriptional repression domains. P. Natl. Acad. Sci. USA. 91, 4509–4513 (1994).
Maeder, M. L. et al. CRISPR RNA-guided activation of endogenous human genes. Nat. Methods 10, 977–979, 10.1038/nmeth.2598 (2013).
Mali, P. et al. RNA-guided human genome engineering via Cas9. Science 339, 823–826, 10.1126/science.1232033 (2013).
Jinek, M. et al. RNA-programmed genome editing in human cells. eLife 2, e00471, 10.7554/eLife.00471 (2013).
Kearns, N. A. et al. Cas9 effector-mediated regulation of transcription and differentiation in human pluripotent stem cells. Development 141, 219–223, 10.1242/dev.103341 (2014).
Gilbert, L. A. et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154, 442–451, 10.1016/j.cell.2013.06.044 (2013).
Hu, J. et al. Direct activation of human and mouse Oct4 genes using engineered TALE and Cas9 transcription factors. Nucleic Acids Res. 42, 4375–4390, 10.1093/nar/gku109 (2014).
Qi, L. S. et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152, 1173–1183, 10.1016/j.cell.2013.02.022 (2013).
Ding, Q. et al. A TALEN genome-editing system for generating human stem cell-based disease models. Cell Stem Cell 12, 238–251, 10.1016/j.stem.2012.11.011 (2013).
Maeder, M. L. et al. Robust, synergistic regulation of human gene expression using TALE activators. Nat. Methods 10, 243–245, 10.1038/nmeth.2366 (2013).
Cermak, T. et al. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 39, e82, 10.1093/nar/gkr218 (2011).
Gao, X. et al. Comparison of TALE designer transcription factors and the CRISPR/dCas9 in regulation of gene expression by targeting enhancers. Nucleic Acids Res. 10.1093/nar/gku836 (2014).
Caffrey, D. R. et al. siRNA off-target effects can be reduced at concentrations that match their individual potency. PloS One 6, e21503, 10.1371/journal.pone.0021503 (2011).
Fedorov, Y. et al. Off-target effects by siRNA can induce toxic phenotype. RNA 12, 1188–1196, 10.1261/rna.28106 (2006).
Lin, X. et al. siRNA-mediated off-target gene silencing triggered by a 7 nt complementation. Nucleic Acids Res. 33, 4527–4535, 10.1093/nar/gki762 (2005).
Holkers, M. et al. Differential integrity of TALE nuclease genes following adenoviral and lentiviral vector gene transfer into human cells. Nucleic Acids Res. 41, e63, 10.1093/nar/gks1446 (2013).
Doyle, E. L. et al. TAL Effector-Nucleotide Targeter (TALE-NT) 2.0: tools for TAL effector design and target prediction. Nucleic Acids Res. 40, W117–122, 10.1093/nar/gks608 (2012).
Strasser, A. et al. Enforced BCL2 expression in B-lymphoid cells prolongs antibody responses and elicits autoimmune disease. P. Natl. Acad. Sci. USA. 88, 8661–8665 (1991).
Acknowledgements
We thank members of the Wu laboratory for valuable discussion. This work was supported in part by a NIDDK/NIH grant (R01DK090478).
Author information
Authors and Affiliations
Contributions
Z.Z. and W.W. designed all of the experiments. Z.Z. and E.W. performed most of the experiments. W.W. supervised the work. Z.Q., E.W., Z.Z. and W.W. wrote the manuscript. All authors reviewed the manuscripts.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Figure S1 and Table S1
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Zhang, Z., Wu, E., Qian, Z. et al. A multicolor panel of TALE-KRAB based transcriptional repressor vectors enabling knockdown of multiple gene targets. Sci Rep 4, 7338 (2014). https://doi.org/10.1038/srep07338
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep07338
This article is cited by
-
Transient Tcf3 Gene Repression by TALE-Transcription Factor Targeting
Applied Biochemistry and Biotechnology (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
