
Abstract
Deposition of amyloid β protein (Aβ) to form neuritic plaques in the brain is the pathological hallmark of Alzheimer's disease (AD). Aβ is produced by β- and γ-cleavages of amyloid β precursor protein (APP). Ubiquitin carboxyl-terminal hydrolase L1 (UCHL1) is a de-ubiquitinating enzyme that cleaves ubiquitin at its carboxyl terminal. Dysfunction of UCHL1 has been reported in neurodegenerative diseases. However, whether UCHL1 affects Aβ production and AD progression remains unknown. Here we report that UCHL1 interacts with APP and regulates Aβ production. UCHL1 increases free ubiquitin level and accelerates the lysosomal degradation of APP by promoting its ubiquitination. Furthermore, we demonstrate that overexpression of UCHL1 by intracranial injection of UCHL1-expressing rAAV reduces Aβ production, inhibits neuritic plaque formation and improves memory deficits in AD transgenic model mice. Our study suggests that UCHL1 may delay Alzheimer's progression by regulating APP degradation in a long-term fashion and that overexpression of UCHL1 may be a safe and effective disease-modifying strategy to treat AD.
Similar content being viewed by others


Introduction
Alzheimer's disease (AD) is the most common neurodegenerative disorder. Aβ, the central component of neuritic plaques, is produced from the amyloid β precursor protein (APP). Under physiological conditions, the majority of APP is processed by α-secretase within the Aβ domain in a non-amyloidogenic pathway. β-cleavage of APP by BACE1 at Asp1 site produces CTFβ C99, which is subsequently processed by the presenilin-dependent γ-secretase complex to generate Aβ in the amyloidogenic pathway1. APP is a type I transmembrane protein and matured in the ER and the Golgi apparatus before reaching the plasma membrane. It is then rapidly internalized to the endosome and sent to lysosome for degradation2,3. Several AD-associated proteins, including BACE1, TMP21, PS1, aph-1 and nicastrin, are degraded by the ubiquitin proteasome system (UPS)4,5,6,7,8. Autophagy-lysosomal degradation may also require ubiquitin signaling, especially when the proteasome is dysfunctional9. While it mainly undergoes lysosomal degradation10,11, APP is also ubiquitinated and degraded by UPS12,13. These findings suggest that ubiquitin proteasome signaling is important for APP processing and Aβ production.
Deubiquitinating enzymes (DUBs) cleave ubiquitin at its terminal carbonyl Gly-76 and the ubiquitin carboxyl-terminal hydrolase L1 (UCHL1) belongs to the UCH family of DUBs. UCHL1 is expressed predominantly in the brain and neuroendocrine systems and accounts for 1–2% of total brain soluble proteins14,15,16. UCHL1 effectively hydrolyzes amino acids from ubiquitin and cleave di-ubiquitins17. It also serves as an ubiquitin ligase at higher concentrations, adding ubiquitin to already mono-ubiquitinated proteins18. In addition, it acts as a free ubiquitin stabilizer, providing ready-to-use ubiquitin for various cellular events19. UCHL1 expression is tightly regulated and NFκB signaling modulates its expression20. Dysfunction of UCHL1 has been reported in many neurodegenerative diseases. The I93M missense mutation in UCHL1 was identified in early-onset familial PD cases21. The E7A recessive mutation resulted in childhood-onset progressive neurodegeneration22. Genetic deletions of UCHL1 in mouse strain, the gad mice, led to phenotypes of gracile axonal dystrophy and premature death23. Reduced UCHL1 protein level was found in sporadic AD brains24 and overexpression of UCHL1 rescued learning and memory deficits in AD model mice by restoring LTP in the hippocampus25. We have shown that UCHL1 regulates BACE1 degradation26. However, whether UCHL1 affects Aβ production and AD progression in a long-term fashion remains unknown.
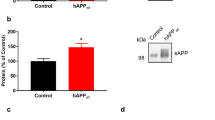
To determine whether UCHL1 regulates APP processing, double transgenic AD model mice APP23/PS45 were injected with AAV1-UCHL1-GFP or control virus at 7 weeks of age. Overexpression of UCHL1 significantly reduced APP CTF production (p<0.01) (Figure 1A, B) and markedly lowered Aβ level in the hippocampi of the UCHL1-overexpressing mice 10 weeks after injection (p<0.05) (Figure 1C). Disruption of UCHL1 gene expression significantly increased APP CTFs in the hippocampi of APP23/gad mice compared to APP23 mice (Figure 1D and E). Furthermore, viral expression of UCHL1 significantly lowered the APP level (p<0.05) (Figure 1F and G) and partial loss of UCHL1 markedly increased the APP level in APP23/gad mice (p<0.05) (Figure 1H, I).

UCHL1 regulates APP processing and Aβ production in vivo.
(A, B) APP CTFs in hippocampal tissues were lower in AAV1-UCHL1-GFP-infected than in AAV1-GFP-infected APP23/PS45 mice controls. N = 8 for each group. **p<0.01 by Student's t-test. (C) Aβ40 of AAV1-UCHL1-GFP-infected mice was significantly lower than control mice. N = 5 for each group. *p<0.05 by Student's t-test. (D, E) APP CTFs from hippocampal tissues of APP23/gad were higher than APP23 mice. N = 8 for each group. *p<0.05 by Student's t-test. (F, G) Endogenous APP from hippocampal tissues of AAV1-UCHL1-GFP-injected was lower than in controls. N = 8 for each group. *p<0.05 by Student's t-test. (H, I) Endogenous APP from hippocampal tissues of APP23/gad were higher than in APP23 mice. N = 8 for each group. *p<0.05 by Student's t-test. The values are expressed as mean ± SEM.
To investigate the underlying mechanisms by which UCHL1 affects APP protein level, the UCHL1 inhibitor LDN-57444 was applied to the Swedish mutant APP cell line 20E2 cells after pZ-UCHL1 transfection (Figure 2A). UCHL1 inhibition led to marked accumulation of mature APP (p< 0.01) (Figure 2B), whereas overexpression of UCHL1 significantly reduced mature APP level in 20E2 cells (p< 0.05) (Figure 2C, D). Similar results were also observed in Haw cell line (Figure 2E and F). Next we examined whether UCHL1 regulates APP ubiquitination/de-ubiquitination process. 20E2 cells were transfected with pZ-UCHL1 and then treated with LDN. Cell lysates were immunoprecipitated with anti-APP C20 antibody followed by immunoblot with anti-ubiquitin antibody. LDN reduced the level of ubiquitinated APP (Figure 2G, upper IP gel). The same membrane was blotted with 9E10 to recognize myc, which was attached to the N-terminus of APP in 20E2 cells. The strongest bands were immature and mature full-length APP (Figure 2G, lower IP gel), which corresponded to APP bands in Figure 2A (at 0 and 6 hours). On the contrary, overexpression of UCHL1 elevated the level of APP-ubiquitin conjugates (Figure 2H). Consistently, overexpression of UCHL1 increased the level of ubiquitinated wildtype APP as well (Figure 2I). The results indicated that UCHL1 accelerated APP degradation by enhancing its ubiquitination.

UCHL1 reduces mature APP protein level and promotes APP ubiquitination.
(A, B) Swedish mutant APP stable 20E2 cells were transfected with pz-UCHL1-st and then treated with LDN. Inhibition of UCHL1 significantly elevated mature APP level. N = 3, **p<0.01 by ANOVA followed by post-hoc Newman-Keuls test. (C, D) Overexpression of UCHL1 markedly reduced mature Swedish mutant APP level in 20E2 cells. N = 3, *p<0.05, by Student's t-test. (E, F) Overexpression of UCHL1 markedly reduced mature wildtype APP level in Haw cells. N = 3, *p<0.05, by Student's t-test. Values represent mean ± SEM. (G) 20E2 cells were transfected with pz-UCHL1-st and then treated with LDN. High-molecular-weight (HMW) APP complex was reduced by LDN. Cell lysates were immunoprecipitated with C20 antibody followed by western blot with anti-ubiquitin antibody. The same membrane was then blotted with 9E10 to recognize myc, which was attached to the N-terminus of APP in 20E2 cells. Negative control lysate was immunoprecipitated with pre-bleed of C20 antibody serum. (H) 20E2 cells were transfected with pz-UCHL1-st or control plasmid for 48 h before lysed. Overexpression of UCHL1 enhanced HMW Swedish APP level. (I) The experiment in (H) was replicated in Haw cells. Overexpression of UCHL1 elevated HMW wildtype APP level.
Facilitation of APP degradation by UCHL1 could be through the lysosomal or proteasomal pathway. We found that lysosomal inhibitor chloroquine significantly accumulated mature APP in the Haw cell (p<0.01) (Figure 3A, B). Surprisingly, proteasomal inhibitor MG132 decreased mature APP level (p<0.05) (Figure 3A, B). It may be due to the blockage of APP maturation by MG13227. Supporting evidence included its little effect on newly synthesized immature APP (lower lane in Fig 3A) but substantial effect on mature APP (upper lane in Fig 3A). The experiments were replicated in 20E2 cells. Chloroquine treatment led to marked APP accumulation (p<0.05) (Figure 3C, D), while MG132 treatment reduced mature APP level (p<0.05) (Figure 3C, D). To investigate whether UCHL1 reduced APP protein level by the lysosomal degradation, Haw cells were transfected with UCHL1 and then treated with 100 μM chloroquine (Figure 3E). Lysosomal inhibition attenuated the effect of UCHL1 on APP protein level, resulting in abolishment of the UCHL1's inhibitory effect on APP (p>0.05) (Figure 3F). We replicated the experiments and obtained same results in 20E2 cells (p>0.05) (Figure 3G and H). Our results indicated that UCHL1 promoted the degradation of APP via the lysosomal pathway.

Lysosomal inhibition attenuates the effect of UCHL1 on APP and UCHL1 interacts with APP and increases free ubiquitin levels in the cell.
(A, B) Haw cells were treated with 5 μM MG132, 100 μM chloroquine or control solutions for 6 h. MG132 treatment reduced mature APP level, whereas chloroquine treatment enhanced it. N = 3, *p<0.05, **p<0.01 by ANOVA followed by post-hoc Newman-Keuls test. (C, D) 20E2 cells were treated MG132, chloroquine or control solutions. N = 3, *p<0.05 by ANOVA. (E, F) Haw cells were transfected with pz-UCHL1-st or control plasmid for 48 h and then treated with chloroquine. Chloroquine treatment attenuated the difference in APP level between UCHL1 and control groups. N = 3, *p<0.05, **p<0.01 by ANOVA. (G, H) The experiments in (E) were replicated in 20E2 cells. N = 3, *p<0.05, **p<0.01 by ANOVA. Values represent mean ± SEM. (I) Haw cells were transfected with pZ-UCHL1-st and then treated with 100 μM chloroquine. Cell lysates were immunoprecipitated with C20 antibody and then immunoblotted with anti-UCHL1 antibody BH7 (Novus Bio). Interaction between UCHL1 and APP was detected and the interaction was enhanced with chloroquine. (J) Haw cells were co-transfected with pZ-UCHL1-st and ubiquitin expression plasmid pCW7. APP was detected by C20 antibody and ubiquitin detected by rabbit anti-ubiquitin antibody (Calbiochem). (K) Overexpression of UCHL1 significantly reduced mature APP level. Overexpression of ubiquitin further downregulated mature APP. N = 3, *p<0.05, **p<0.01 by ANOVA. (L) Overexpression of UCHL1 upregulated ubiquitin level. N = 3, *p<0.05, **p<0.01 by ANOVA. Values represent mean ± SEM.
To investigate whether UCHL1 physically interacts with APP, Haw cells were transfected with UCHL1 and then treated with chloroquine. Co-immunoprecipitation assay showed the interaction between UCHL1 and APP and the interaction was potentiated by chloroquine (Figure 3I). To investigate whether UCHL1 promotes the ubiquitination and degradation of APP by regulating the free ubiquitin pool, Haw cells were co-transfected with pZ-UCHL1-st and ubiquitin-expression plasmid pCW7. Overexpression of UCHL1 significantly elevated free ubiquitin levels (p<0.05) (Figure 3J, L) and overexpression of ubiquitin further increased free ubiquitin levels (p<0.01) (Figure 3J, L). Mature APP level was reduced by UCHL1 as expected (p<0.05) (Figure 4J) and was further reduced by ubiquitin expression (p<0.01) (Figure 3J, K). The results indicate that the increased free ubiquitin level by UCHL1 may be responsible for increasing ubiquitinated APP and decreasing mature APP.

Overexpression of UCHL1 decreases plaque formation and improves learning and memory deficits.
(A) Viruses were bilaterally injected into the hippocampal region of APP23/PS45 mice at the age of seven weeks old. The mice were then sacrificed at 17 weeks old. Neuritic plaques were detected by 4G8 antibody. The arrows indicate plaques in AAV1-GFP control virus (Aa) or AAV1-UCHL1-GFP injection (Ab). The plaques were confirmed by thioflavin-S staining (Ac and d). (B) Quantification of neuritic plaques in (A, a-b) by Image J (NIH). The value represents mean ± SEM. n = 8 for each group. **p < 0.01 by Student's t-test. (C) Detection of neuritic plaques in the cortical region in AAV1-UCHL1-GFP -injected mice (Cb) and controls (Ca). The plaques were confirmed by thioflavin-S staining (Cc and d). Bar: 500 μm. (D) Quantification of neuritic plaques in (C, a-b). n = 8 for each group. **p < 0.01 by Student's t-test. (E) Neuritic plaques in six-month-old APP23 and APP23/gad mice were examined by thioflavin-S staining. (F) Quantification of (E). n = 8 for each group. *p < 0.05 by Student's t-test. (G-J) Eight weeks after AAV1-UCHL1-GFP or AAV1-GFP injection, APP23/PS45 mice were subjected to Morris water maze test at the age of 15 weeks old. n = 16 for AAV1-GFP group, n = 13 for AAV1-UCHL1-GFP group. On the first day of visible platform test, AAV-UCHL1-GFP-injected mice displayed similar escape latency (G) and swimming speed (H) as control mice. p>0.05 by Student's t-test. (I) During hidden platform test, AAV1-UCHL1-GFP mice exhibited shorter escape latency. *p<0.05 by two-way ANOVA group comparison. (J) On day 6 of the probe trial, AAV1-UCHL1-GFP-injected mice spent more time in the target quadrant than controls. *p<0.05 by Student's t-test. The values are expressed as mean ± SEM. (K-N) Six-month-old APP23 or APP23/gad mice were subjected to Morris water maze test. n = 12 for APP23 group, n = 11 for APP23/gad group. (K) Escape latency and (L) swimming speed. P>0.05 by Student's t-test. (M) Hidden platform tests. *p<0.05 by two-way ANOVA post-hoc Bonferroni test. (N) The probe trial on day 6. *p<0.05 by Student's t-test.
Next we examined the effect of UCHL1 on Alzheimer's development in vivo. APP23/PS45 mice were intracranially injected with AAV1-UCHL1-GFP or its control AAV1-GFP at 7 weeks of age. GFP and UCHL1 protein level were markedly increased in UCHL1-GFP virus-infected hippocampal area 10 weeks after injection (Supplemental Figure 1B and C). In mice that were unilaterally injected with AAV1-UCHL1-GFP, GFP was robustly expressed in dentate gyrus and CA3 of the hippocampus ipsilaterally (Supplementary Figure 1D). In addition, UCHL1 was co-expressed with GFP in AAV1-UCHL1-GFP-infected mice, but not in those infected with AAV1-GFP (Supplemental Figure 1E). UCHL1 overexpression significantly reduced the number of neuritic plaques in the hippocampal region (p<0.01) (Figure 4Aa and b, B) and in the neocortex area of APP23/PS45 mice (p<0.01) (Figure 4Ca and b, D). Thioflavin-S staining confirmed the reduction of plaque formation in UCHL1-overexpressed mice (Figure 4Ac,d and Cc, d). We also knocked down UCHL1 expression in APP23 transgenic mice by breeding APP23 mice with heterozygous gad mice. At 6 months old APP23 mice merely started to develop Aβ plaques, with at most 1 to 2 plaques per slice. The plaque numbers were increased in APP23/gad mice compared to APP23 (Figure 4E and F). The results indicate that knockdown of UCHL1 expression facilitates neuritic plaque formation in APP23 mice.
To investigate whether UCHL1 overexpression improves learning and memory deficit in AD model mice, Morris water maze was carried out in AAV-UCHL1-GFP-injected APP23/PS45 mice and the control group. There were no difference of the escape latency (p>0.05) (Figure 4G) or swimming speed (p>0.05) (Figure 4H) on day 1 of the visible platform test between the UCHL1-overexpressed and control groups, indicating that viral infection and UCHL1 overexpression did not affect the swimming ability or vision. In the hidden platform test UCHL1-overexpression significantly shortened escape latency of the AD mice (p<0.05) (Figure 4I). In the probe trial test, UCHL1-overexpressing mice spent markedly more time in the hidden platform quadrant (p<0.05) (Figure 4J). To further confirm UCHL1's effect on learning and memory deficits, we crossed the gad mice with APP23. There were no difference of the escape latency (p>0.05) (Figure 4K) or swimming speed (p>0.05) in the visible platform test (Figure 4L) between APP23 and APP23/gad mice. In the hidden platform tests, APP23/gad mice showed significantly prolonged escape latency compared to APP23 mice (p<0.05) (Figure 4M). APP23/gad mice passed the virtual platform less frequently than APP23 mice in the hidden platform test (p< 0.05) (Figure 4N). Taken together, these data demonstrated that UCHL1 expression rescued memory deficits in AD model mice, whereas reducing UCHL1 expression exacerbated the memory deficits in AD model mice.
Dysfunction of UCHL1 has been implicated in neurodegenerative diseases. Here we demonstrated that UCHL1 interacts with APP and that APP undergoes UCHL1-assisted ubiquitination followed by trafficking to lysosome for degradation. Our study showed that reduction of UCHL1 exacerbates AD-like pathology and behavioral performance in APP23/gad mice. The data suggests that the lower expression of UCHL1 may be partially responsible for Alzheimer pathophysiology and cognitive impairment. We delivered UCHL1 into the hippocampal region of the mice by intracranial injection of UCHL1-expressing AAV. Overexpression of UCHL1 reduced Aβ production, inhibited neuritic plaque formation and improved memory deficits in AD model mice. Long-term expression by AAV delivery has been verified with no significant adverse events from the viral delivery method. Our work is the first to suggest that UCHL1 delays AD progression in a long-term fashion and that rAAV-mediated UCHL1 gene therapy to overexpress UCHL1 in the brain could be a promising disease-modifying strategy for AD therapeutics.
Methods
Experimental procedures were carried out in accordance with the guidelines and approved protocols by The University of British Columbia Animal Care and Use Committee and Biosafety Committee. To generate the adeno-associated virus (AAV) expressing UCHL1, the human UCHL1 cDNA sequence was cloned into the pAAV-GFP-cDNA6 vector. APP23 transgenic mice carry human APP751 cDNA with the Swedish mutation and PS45 transgenic mice carry human PS1 cDNA with the G384A mutation. The gracile axonal dystrophy (gad) mouse carries a spontaneous mutant with an in-frame deletion in exons 7 and 8 of UCHL1 and is equivalent to a UCHL1 knockout mouse model. APP and Aβ proteins were assayed in AAV-infected AD mice or APP23/gad mice. Neuritic plaque formation was assessed by immunostaining with 4G8 antibody and Thioflavin-S staining28,29. Morris water maze test was used to determine the learning and memory deficits30. Full Methods and any associated references are described in Online Supplemental Data.
References
Deng, Y. et al. Amyloid-beta protein (Abeta) Glu11 is the major beta-secretase site of beta-site amyloid-beta precursor protein-cleaving enzyme 1(BACE1) and shifting the cleavage site to Abeta Asp1 contributes to Alzheimer pathogenesis. Eur J Neurosci 37, 1962–1969 (2013).
Koo, E. H., Squazzo, S. L., Selkoe, D. J. & Koo, C. H. Trafficking of cell-surface amyloid beta-protein precursor. I. Secretion, endocytosis and recycling as detected by labeled monoclonal antibody. J Cell Sci 109 (Pt 5), 991–998 (1996).
Small, S. A. & Gandy, S. Sorting through the cell biology of Alzheimer's disease: intracellular pathways to pathogenesis. Neuron 52, 15–31 (2006).
Qing, H. et al. Degradation of BACE by the ubiquitin-proteasome pathway. Faseb J 18, 1571–1573 (2004).
He, G. et al. Ubiquitin-proteasome pathway mediates degradation of APH-1. J Neurochem 99, 1403–1412 (2006).
He, G. et al. Degradation of nicastrin involves both proteasome and lysosome. J Neurochem 101, 982–992 (2007).
Fraser, P. E. et al. Presenilin 1 is actively degraded by the 26S proteasome. Neurobiol Aging 19, S19–21 (1998).
Liu, S. et al. TMP21 degradation is mediated by the ubiquitin-proteasome pathway. Eur J Neurosci 28, 1980–1988 (2008).
Kraft, C., Peter, M. & Hofmann, K. Selective autophagy: ubiquitin-mediated recognition and beyond. Nat cell biol 12, 836–841 (2010).
Caporaso, G. L., Gandy, S. E., Buxbaum, J. D. & Greengard, P. Chloroquine inhibits intracellular degradation but not secretion of Alzheimer beta/A4 amyloid precursor protein. Proc Natl Acad Sci U S A 89, 2252–2256 (1992).
Haass, C., Koo, E. H., Mellon, A., Hung, A. Y. & Selkoe, D. J. Targeting of cell-surface beta-amyloid precursor protein to lysosomes: alternative processing into amyloid-bearing fragments. Nature 357, 500–503 (1992).
Watanabe, T., Hikichi, Y., Willuweit, A., Shintani, Y. & Horiguchi, T. FBL2 regulates amyloid precursor protein (APP) metabolism by promoting ubiquitination-dependent APP degradation and inhibition of APP endocytosis. J Neurosci 32, 3352–3365 (2012).
Kaneko, M. et al. Loss of HRD1-mediated protein degradation causes amyloid precursor protein accumulation and amyloid-beta generation. J Neurosci 30, 3924–3932 (2010).
Doran, J. F., Jackson, P., Kynoch, P. A. & Thompson, R. J. Isolation of PGP 9.5, a new human neurone-specific protein detected by high-resolution two-dimensional electrophoresis. J Neurochem 40, 1542–1547 (1983).
Wilson, P. O. et al. The immunolocalization of protein gene product 9.5 using rabbit polyclonal and mouse monoclonal antibodies. Br J Exp Pathol 69, 91–104 (1988).
Jackson, P. & Thompson, R. J. The demonstration of new human brain-specific proteins by high-resolution two-dimensional polyacrylamide gel electrophoresis. J Neurol Sci 49, 429–438 (1981).
Larsen, C. N., Krantz, B. A. & Wilkinson, K. D. Substrate specificity of deubiquitinating enzymes: ubiquitin C-terminal hydrolases. Biochemistry 37, 3358–3368 (1998).
Liu, Y., Fallon, L., Lashuel, H. A., Liu, Z. & Lansbury, P. T., Jr The UCH-L1 gene encodes two opposing enzymatic activities that affect alpha-synuclein degradation and Parkinson's disease susceptibility. Cell 111, 209–218 (2002).
Osaka, H. et al. Ubiquitin carboxy-terminal hydrolase L1 binds to and stabilizes monoubiquitin in neuron. Hum Mol Genet 12, 1945–1958 (2003).
Wang, R. et al. NF-kappaB signaling inhibits ubiquitin carboxyl-terminal hydrolase L1 gene expression. J Neurochem 116, 1160–1170 (2011).
Leroy, E. et al. The ubiquitin pathway in Parkinson's disease. Nature 395, 451–452 (1998).
Bilguvar, K. et al. Recessive loss of function of the neuronal ubiquitin hydrolase UCHL1 leads to early-onset progressive neurodegeneration. Proc Natl Acad Sci USA 110, 3489–3494 (2013).
Yamazaki, K. et al. Gracile axonal dystrophy (GAD), a new neurological mutant in the mouse. Proc Soc Exp Biol Med 187, 209–215 (1988).
Choi, J. et al. Oxidative modifications and down-regulation of ubiquitin carboxyl-terminal hydrolase L1 associated with idiopathic Parkinson's and Alzheimer's diseases. J biol chem 279, 13256–13264 (2004).
Gong, B. et al. Ubiquitin hydrolase Uch-L1 rescues beta-amyloid-induced decreases in synaptic function and contextual memory. Cell 126, 775–788 (2006).
Zhang, M. et al. Control of BACE1 degradation and APP processing by ubiquitin carboxyl-terminal hydrolase L1. J Neurochem 120, 1129–1138 (2012).
Steinhilb, M. L., Turner, R. S. & Gaut, J. R. The protease inhibitor, MG132, blocks maturation of the amyloid precursor protein Swedish mutant preventing cleavage by beta-Secretase. J Biol Chem 276, 4476–4484 (2001).
Qing, H. et al. Valproic acid inhibits Abeta production, neuritic plaque formation and behavioral deficits in Alzheimer's disease mouse models. J Exp Med 205, 2781–2789 (2008).
Sun, X. et al. Hypoxia facilitates Alzheimer's disease pathogenesis by up-regulating BACE1 gene expression. Proc Natl Acad Sci U S A 103, 18727–18732 (2006).
Ly, P. T. et al. Inhibition of GSK3beta-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J Clin Invest 123, 224–235 (2013).
Acknowledgements
This work was supported by Canadian Institutes of Health Research (CIHR) Grant IAP-102225, MOP-97825 and TAD-117948 and Jack Brown and Family Alzheimer's Research Foundation. W.S. was the holder of the Tier 1 Canada Research Chair in Alzheimer's Disease. M. Z. and Si Z. are supported by UBC 4YF Scholarship. Shuting Z. was supported by the Chinese Scholarship Council award.
Author information
Authors and Affiliations
Contributions
M.Z. and W.S. conceived and designed the experiments; M.Z., F.C., Sh.Z. and Si.Z. performed the experiments; M.Z. and W.S. analyzed and contributed reagents/materials/analysis tools; M.Z. and W.S. wrote the paper. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Online supplementary methods and figure
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Zhang, M., Cai, F., Zhang, S. et al. Overexpression of ubiquitin carboxyl-terminal hydrolase L1 (UCHL1) delays Alzheimer's progression in vivo. Sci Rep 4, 7298 (2014). https://doi.org/10.1038/srep07298
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep07298
This article is cited by
-
Alterations of mRNAs and Non-coding RNAs Associated with Neuroinflammation in Alzheimer’s Disease
Molecular Neurobiology (2024)
-
Regulatory mechanisms and therapeutic potential of JAB1 in neurological development and disorders
Molecular Medicine (2023)
-
Potential diagnostic value of CSF metabolism-related proteins across the Alzheimer’s disease continuum
Alzheimer's Research & Therapy (2023)
-
The Emerging Roles of E3 Ligases and DUBs in Neurodegenerative Diseases
Molecular Neurobiology (2023)
-
Treadmill exercise promotes E3 ubiquitin ligase to remove amyloid β and P-tau and improve cognitive ability in APP/PS1 transgenic mice
Journal of Neuroinflammation (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
