
Abstract
This study demonstrates both prokaryotic and eukaryotic community structures and dominant taxonomies in different positions of the greatest estuary reservoir for drinking water source in the world in four seasons of one year using 454 pyrosequencing method with total of 312,949 16S rRNA and 374,752 18S rRNA gene fragments, including 1,652 bacteria OTUs and 1,182 fungus OTUs. During winter and spring, the community composition at the phylum level showed that microorganisms had similar structures but their quantities were different. Similarly, obvious changes at the genus level were observed among the samples taken in winter and spring between summer and fall. Microorganisms located the reservoir inlet were founded to be different from those in rear at both phylum and genus level. Air temperature had a stronger effect than sampling location on the microbial community structure. Total nitrogen and dissolved oxygen were algae-monitoring indicators during the whole year. Moreover, Bacillus was an efficient indicator during summer and autumn for bacteria OTUs.
Similar content being viewed by others


Introduction
Estuary ecosystems are important water sources for numerous cities, particularly world-class, densely populated metropolitan areas located at the junction of river and ocean, such as New York, London, Tokyo, Singapore, Hong Kong and Shanghai1,2,3,4. Microbial communities are a key constituent of the microbial food loop of estuary reservoirs, being a decomposer, consumer, primary producer and secondary producer. Water quality is strongly influenced by microbial community dynamics and ecosystem functions such as organic matter content and nutrient recycling5. Moreover, these changes in the microbial community trigger algae blooms and off-flavor problems, which negatively affect the water supply. Understanding the community composition and time-site distribution of estuary reservoir microorganisms increases the current understanding of the distribution characteristic and its function in the estuarine ecosystem and is particularly helpful in judging the environmental safety of the water supply as a source of drinking water.
Microscopic identification and plate cultivation6 are used to monitor and characterize the microbial diversity of aquatic ecosystems, but these two methods cannot differentiate uncultured species. Some biochemical methods, such as phospholipid fatty acids (PLFA) analysis7, are used to characterize the community structure of microbiota, including both prokaryotes and eukaryotes. However, PLFA analysis has certain limitations, such as overlap in the composition of microorganisms. Both denaturing gradient gel electrophoresis fingerprinting (DGGE) and clone library analysis8 have made independent cultivation possible. However, these tools usually underestimate the overall diversity of a microbial community and are difficult to use to obtain a comprehensive community profile in complex environments. Fortunately, pyrosequencing, a high-throughput sequencing technology, has gained increasing research interest as a novel tool for examining community structure9,10. Moreover, several researchers11 showed a general comparison about four basic approaches: miTags, m454Tags, 454Tags and iTags, advocated the technique-miTags (metagenomic illumina 16S tags) to acquire a large amount of funcional data and avoid a mass of PCR biases. However, many difficulties existed in metagenomic analysis due to variety of genes, variance at evolutionary rates, complex in computations, especially absence of appropriate reference gene sets. Therefore, 454 Pyrosequencing are more fit for rRNA diversity analyses.
Similar to prokaryotes, including eukaryotic algae, fungus and macrophytes have also been closely associated with water environment problems, such as algae bloom and off-flavor of freshwater. Principal component analysis (PCA) has shown that phytoplankton could be an indicator of improved water quality at the Golden Horn Estuary12. Certain fungi species have been associated with the production of the mycotoxin patulin and the odor secondary metabolite13. A study by Kravehuk et al14 suggested that macrophytes were a positive factor in bloom development and contributed to the accumulation of akinetes in littoral sediments. Although scientific interest in structure and the function of eukaryotic microbes has increased, numerous studies5,15 have largely focused on bacteria. Minimal differences in environmental conditions influence the composition and function of microbial communities in water resources and environmental heterogeneity caused by different sites appear to be the key factor affecting the biomass of phytoplankton16. Location and season could significantly affect the partial concentrations of nitrogen and phosphorus, thereby affecting community composition. Several studies17,18 have been conducted to characterize the structures of various space and time conditions in aquatic systems, whereas the effect of overall diversity on phytoplankton has never been addressed. In the present study, an estuary reservoir with different environmental conditions was selected using time-site treatments and pyrosequencing was used to characterize whole communities. The results could provide new insight into the relationship between species and environmental conditions in estuary reservoirs to benefit water source protection research. Understanding both prokaryotic and eukaryotic community structure is also necessary to better understand microbial ecology and potential algae outbreaks in water sources.
Results
Environmental parameters
Supporting Information (SI) Table S1 lists the environmental parameters, including water temperature, dissolved oxygen (DO), permanganate index (CODMn), total nitrogen (TN), total phosphorus (TP), transparency and chlorophyll-a, microcystin-RR (MC_RR) and microcystin-LR (MC_LR). Transparency increased whereas TP decreased from the head to the tail of the reservoir (Fig. 1). CODMn and chlorophyll-a concentrations were higher in Site 3 than in Site 2 (Fig. 1). The chlorophyll-a of Site 0 was always lower than those of other sites and dramatically influenced by air temperature as well as CODMn, particularly by air temperatures ranging from 22°C to 30°C and water quality that meets the current national “Sanitary standard for drinking water” (GB5749-2006). Water temperature varied more significantly in periods. Irregular changes in TN were observed in the six sites. The DO of all samples were relatively stable at approximately 9.11 mg/l. MC_RR and MC_LR were lower than 0.5 μg/l in most samples, with only the MC_LR in sample 0410-3 reaching 0.77 μg/l, which was still lower than the national standard of 1 μg/l.

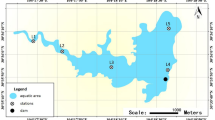
Locations and sampling sites in estuary reservoir.
Six monitoring positions were marked as 0, 2, 3, 8, 13, 15 respectively. Satellite image was obtained by synthesizing the data from Satellite Environment Center, Ministry of Environmental Protection in China in free of charge in website of [http://www.secmep.cn/secPortal/portal/column/itemDetails.faces?itemid=8ae67f88272ce22801272d0f50410005] using commercial GeoStar3.0 (http://www.geostar.com.cn) and free Gimp 2.8.10 software (http://www.gimp.org/).
Overall taxonomic richness
A total of 312,949 16S rRNA sequences and 374,752 18S rRNA sequences were selected for classification. Dominant length distributions were approximately 429 and 420 bp, respectively. The taxonomic distributions of the 16S rRNA sequences obtained in the water samples are shown in Fig. 2a. The majority of bacteria sequences (105,673 sequences) belonged to Cyanophyta. The second largest group was Actinobacteria (49,298), followed by Firmicutes (40,013). Betaproteobacteria and Alphaproteobacteria were the most abundant Proteobacteria, making up approximately 56% and 30%, respectively, of the Proteobacteria sequences (43,091). Based on the 97% species similarity, a total of 1,652 OTUs derived from 280,382 sequences were detected in this study. Among these OTUs, 309 were Bacteroidetes OTUs (18.7%), 233 were Cyanophyta (14.1%) and 198 were Alphaproteobacteria (12.0%). The taxonomic distributions of the 18S rRNA sequences obtained in the water samples are shown in Fig. 2b. The majority of fungal sequences (129,819 sequences) belonged to Metazoa. The second largest group was Cryptomonadales (72,845), followed by Chlorophyta (30,194). Eukaryotic algae included Cryptomonadales, Chlorophyta, Diatomea, Dinoflagellata, Kathablepharidae, Chrysophyceae, Glaucophyta, Stramenopiles and Prymnesiophyceae (126,631 sequences, accounting for 36.4% of total sequences). Similarly, a total of 1,182 OTUs derived from 357,732 fungal sequences were detected in this study. Among these OTUs, 177 were Cercozoa OTUs (15.0%), 129 were Chlorophyta (11.0%), 93 were Metazoa (7.9%), 53 were Cryptomonadales (4.5%) and 27 were Diatomea (2.3%).

(a–b) Different taxonomic numbers of sequence and derived OTU in prokaryote and eukaryote.
Effects of season-site treatments at phylum levels on community
Shifts in the proportions of sequences for different bacteria phyla are shown in Fig. 3a. In general, different samples showed a relative similar trend in terms of richness as well as indicated by Chao1 and Ace indexes during the same season (S2a). The composition and biomass of taxa exhibited a remarkable spatial and temporal heterogeneity with a pronounced longitudinal gradient. Cyanophyta, Proteobacteria, Actinobacteria, and Bacteroidetes were always highest across all water samples, with more than 80% proportion. However, Sites 0 and 2 exhibited greatly decreased relative abundance of Proteobacteria in summer and autumn, as shown in green type. Except for autumn, the proportion of Cyanophyta in Sites 0 and 2 were well below those of other sites. Firmicutes proportions were highest in the summer samples. Changes in the proportion of sequences of different fungal phyla are shown in Fig. 3b and richness in Apring and Summer significantly higher than others (S2b). Interestingly, except for the winter samples, Metazoa had the highest abundance among the samples. Cryptomonadales always existed in all samples and were highest in number in spring and autumn. Cercozoa were most plentiful during winter, particularly in Sites 3, 8 and 13, accounting for more than 60% in those sites, such a high percentage was associated with low frequency during winter sampling. Chlorophyta had significantly stronger samples in summer than others, similar to Diatomea.

(a–b) Phylum-level changes in different season-site assemblages of prokaryote and eukaryote sequences. The phylum level distribution presented is based on the 80% similarity clusters of OTUs.
Effects of season-site treatments at genus level on abundance and taxonomy
The hierarchical heatmap was based on the top 50 abundant bacterial communities at the genus level, which were classified into four groups (Fig. 4a). The first group was mainly composed of the summer samples, the second group was composed of the autumn samples, the third group was composed of the spring and winter samples from the middle-rear parts of the reservoir and the last group was composed of the spring and winter samples from the front-middle parts. The most abundant bacteria at genus level were Chloroplast norank (22.8%), hgcI clade (11.8%), Bacillus (7.6%), Synechococcus (7.0%) and Microcystis (2.4%). Microcystis were most abundant in summer at Sites 3 and 8 and Microcystis aeruginosa accounted for only 3.9% of the bacteria. Moreover, Synechococcus always found during summer and fall. Given the same seasons for all of the samples, such as for the summer and autumn samples that were still clustered together, the features of the top 50 fungus genera were less consistent than those of the bacteria (Fig. 4b). The most abundant fungi were Cryptomonas (16.5%), Maxillopoda (13.5%), Mammalia_norank (12.4%), Novel_Clade_2_norank (4.0%) and Rotifera_norank (9.7%). Teleaulax and Chlamydomonas were the three most abundant eukaryotic algae. Furthermore, algae such as Closterium, Chlorophyceae norank, Hafniomonas, Cyclotella, Actinocyclus and Ceratium, increased sharply during summer, whereas Thalassionsira were not found in the summer samples.

(a–b) Heatmap analysis of prokaryote and eukaryote communities based on the Bray-Curtis similarity of 454 pyrosequencing profiles.
Statistical characterization of complete pyrosequencing data obtained from estuary reservoir
Statistical analysis of 16S rRNA showed that because of the different time-site treatments, environmental variables and bacterial characteristics were different between seasons and stations, particularly among the dominant species (Fig. 5a). The total eigenvalues in the 16S rRNA data set accounted for 2,687,740. The total eigenvalues explained by environmental eigenvalues accounted for 779,902, occupying 29.0% of the total RDA explanatory power. Concerning the variation of species data, the first axis explained 55.1%, the first and second axes together explained 76.7%. The first axis explained 16.5% of the variation of species-environment relation, the first and second axes together explained 26.1%. Biplot scaling of redundancy discriminate analysis (RDA) with original data were shown in Fig. 5a and S3a. Both Temperature and MC_RR got 0.001 Pr with 999 number of permutations based on the 1‰ level in a permutation test for all constrained eigenvalues for prokaryotes. Corresponding values were 0.002 for TN and 0.01 for DO and greater than 0.05 for transparency, CODMn, etc. Therefore, four environmental factors, Temperature, MC_RR, TN and DO, were significant environmental variation for bacteria. Moreover, it made a statistically significant contribution from Bacillus to the species-environment relationship (P = 0.001, F = 2.9513, number of permutations = 1,000). The higher contents of TN and CODMn as well as increase in chlorophyll-a and Bacillus and in activity were observed for partial samples from Groups 4 and 5 (0828, 0911 and 0913) when the air temperature during sampling decreased from 30.5°C to 28.8°C. The increase in Synechococcus related to activity was observed for partial samples of Groups 3 and 4 (0621, 0712, 0725 and 0807). However, the lower air temperature of samples resulted in higher DO values as well as growth in MC_LR. Compared with the effect of sites, these observations indicated that different air temperatures drastically changed the bacterial community structure. Given the rapid growth of Microcystisaeruginosa when air temperature decreased from 30.5°C to 28.8°C, researchers should pay more attention to monitoring indices such as Temperature, MC_RR and TN or the sudden abnormal appearance of Bacillus.

(a–b) Redundancy discriminant analysis (RDA) biplot of the distribution of prokaryote and eukaryote communities with environmental variables in estuary reservoir. The upper right legend shows color-coding indicating six groups in estuary reservoir. group 1: 0226, group 2: 0328, 0410, 0426, group 3: 0529, 0621, group 4: 0712, 0725, 0807, 0828, group 5: 0911, 0913, 1011, group 6: 1029.
The statistical analysis of 18S rRNA indicated that the summer and fall samples were located at the right side of the PC2 axes, whereas the winter and spring samples were scattered at the left side (Fig. 5b). Correspondingly the total eigenvalues in the 18S rRNA data set accounted for 4,104,485. The total eigenvalues explained by environmental eigenvalues accounted for 924,151 and the combination of these environmental factors provided an additional 22.5% of the total RDA explanatory power. Concerning the variation of species data, the first axis explained 45.1%, the first and second axes together explained 66.8%. The first axis explained 11.0% of the variation of species-environment relation, the first and second axes together explained 15.3%. Biplot scaling of RDA with original data were shown in Fig. 5b and S3b. Temperature and DO got 0.001 Pr with 999 number of permutations based on 1‰ level in a permutation test for all constrained eigenvalues for eukaryotes. In addition, none of the other factors made a statistically significant contribution to the species-environment relationship (P = 0.173, F = 1.1948, number of permutations = 1000). During the rapid growth of Microcystisaeruginosa, TP was related to Cryptomonas and partial samples from Groups 4 and 5, which showed a relative important contribution of TP to the activity of eukaryotic microorganisms. Data revealed that Actinocyclus and Cryptomonas were highly abundant. Cryptomonas accounted for 16.5% of the 18S rRNA sequences, indicating that taxonomy was a key factor in the algae community throughout the year. Rapid proliferation of algae during spring and winter were mainly caused by eukaryotic algae, whereas the activity of Microcystis was associated with late summer and early fall. Thus, more attention should be given to Temperature and DO when monitoring algae in winter and spring.
Discussion
The Changjiang Estuary was an important interface between terrestrial and marine environments and also had highly complex and dynamic because of the input of freshwater and seawater alternately. The study on temporal and spatial dynamics of prokaryotic and eukaryotic microbes throughout the whole year by pyrosequencing is very important for understanding the structure and function of the ecosystem for estuarine reservoir. Given the supposedly universal primers and database discrepancy, a number of sequences belonging to taxa different from Cyanophyta and other unclassified organisms were detected (Fig. 2a,b). Nevertheless, nearly 105,673 Cyanophyta sequences were obtained, accounting for 37.7% of the bacterial sequences and forming 233 OTUs. The results were similar to other pyrosequencing study19 on aquatic ecosystems, that is, the dominant bacteria were Cyanophyta, accounting for 32.3% of the total sequences in the Guanting Reservoir. This study the eukaryotic algae accounted for 36.4% of fungal sequences, Cryptomonas were the most abundant genus (16.5% of fungal). The results are consistent with those of the other methods18,20, that is, phytoplankton were the dominant taxa in a reservoir or a lake. In addition, these discussed results conflicted with previous report21 that most of the OTUs belonged to Actinobacteria, followed by Bacteroidetes and Betaproteobacteria by using the restriction fragment length polymorphism (T-RFLP). The discrepancy between the two reservoirs were mainly caused by the higher percentage of uncultured clones by using different methods except for regional difference. Microbial community diversity was traditionally based on morphology or physiology. 16 different algae by observing phytoplankton biovolume in a subtropical estuary22. A total of 142 phytoplankton taxa were identified from eight algae classes during four years in the Golden Horn Estuary23 using quantitative analysis of phytoplankton, which were significantly less than the 233 prokaryotic algae OTUs and the 209 eukaryotic algae OTUs found by pyrosequencing. Since 1993, DGGE has been employed to study microbial community composition24. DGGE isolated 101 sequences and 46 clones and DNA-based fingerprinting obtained 45 16S rRNA OTUs and 39 18S rRNA OTUs17. However, compared with the traditional molecular biology methods, the pyrosequencing used in this study obtained 312,949 16S rRNA sequences and 374,752 18S rRNA sequences. These results indicate that the numbers of derived sequences were enough to characterize community diversity and richness and provide more detailed information about uncultured taxa in the estuary reservoir. In addition, the profile of whole community was often reported by only one type25,26 or several types3,17 of species in previous estuary studies. Species compositions known in estuary ecosystems were much less those of terrestrial8,10 and marine15,27 ecosystems. The use of pyrosequencing27,28 has proven to be one of the most effectively techniques to determine the identity and to quantify the abundance of microorganisms, as well as to reveal the profile of whole community accurately in natural system. Hence, the long time pyrosequencing results of reservoir had important values for similar reservoir.
Although the prokaryotic community was dominated by Cyanobacteria, Chloroplast norank at the genus level accounted for the majority of Cyanobacteria in spring. These groups were not found using techniques in microscopy and were not the study subject in the Cyanobacteria bloom. The eukaryotic community was dominated by Cryptomonas (accounting for 16.5% of sequences). Although Edward et al22 reported that Cyanobacteria, Diatoms and Dinoflagellate were the dominant algae in an aquatic ecosystem. These results were in accordance with the observation of Pavel et al29 that Crytophyte and Diatom prevailed in the reservoir from April to mid-June. These complicated categorical information on microbial communities extend the knowledge obtained from previous studies and reports. At a higher resolution, Synechococcus and Cryptomonas were the dominant genera among Cyanobacteria and Cryptophyta, respectively. Synechococcus survive and propagate more easily than the others because of the main participants in the global carbon cycle and the primary contributors to primary productivity and are particularly abundant in near shore waters. Cryptomonas widely exist because of their ability to adapt to a wide range of irradiance and their high tolerance for eutrophication30. Remarkably, a large number of environmental and unclassified Chloroplast norank were found in estuary reservoirs, which is consistent with the finding of Zeng et al31 that most Cyanobacteria were related to chloroplasts (93.7%). The developments of both pyrosequencing and microbial taxonomy can provide the basis for estuary reservoirs to be a potential water source for a megalopolis. A more interesting study32 indicated that the relationship of phylogeny and function was sufficiently linked that “predictive metagenomic” approach, which provided useful insights into those uncultivated microbial communities such as “no rank species” by using marker gene data. Aside from Synechococcus, Bacillus and Microcystis, hgcI clade and Limnohabitans were also abundant in the bacterial sequences. HgcI clade belonged to Actinomycetales and previous case33 reported that such microbes were affected by the odor and taste of the drinking water. Limnohabitans belong to Betaproteobacteria and Kasalicky et al34 suggested that Limnohabitans had a prominent role in freshwater bacterioplankton communities because of their high rates of substrate uptake and growth, growth on algae-derived substrates and high mortality rates from bacterivory. The most abundant groups among the eukaryotes were Maxillopoda, Mammalia_norank, Rotifera_norank and Novel_Clade_2_norank (close relationship with Cercomonas). Teleaulax, Chlamydomonas and Actinocyclus were also abundant. Such rich algae sequences were due to troubles in these potential water sources largely caused by phytoplankton and occupied a high biomass in freshwater aquatic systems. This study has identified the key genera involving in abnormal proliferation of algae or off-flavor problems from water source region, also determined their respective roles in aquatic ecological system.
Microorganism community compositions in the estuary23,28 were reported with little knowledge about the effect of environmental factors on the diversity and distribution of microorganism. RDA was powerful27,29 in determining the relationship between microbiological community composition and environmental factors. Previous studies23,31,35 demonstrated that temperature and nutrients were the key factors in the composition of aquatic communities and that DO, CODMn and chlorophyll-a were the main direction indices for algae growth. Thus, this study selected six abiotic environmental indices (water temperature, transparency, DO, CODMn, TP and TN) as well as biotic environmental indices (Chlorophylla-a, MC-RR and MC-LR concentrations, five predominant sequences). Pyrosequencing studies not only provide an insight into the microbial assemblages in estuary reservoirs but also determine their ecological function through the shifts in their compositions under various time-site treatments. Compared with the estuary reservoirs used in other studies35,36, the one used in this study had acceptable levels of chlorophyll-a and microcystin risk during the entire summer (average 6.41 μg/l, 0.12 of MC-RR and 0.05 MC-LR μg/l). Environmental conditions were varied among the time-site treatments so that the communities varied as well, thereby increasing the heterogeneity of microbial communities, in accordance with the findings of Yan et al17. Samples from 0 and 2 denoted the inlet and start site of the reservoir, respectively (Fig. 2). Heterogeneity of environmental conditions resulted in the minimum number of Cyanophyta and maximum number of Proteobactria in different monitored sampling sites. High abundance of Proteobactria was observed in a previous study37. Heterogeneity was also reported from the similar estuary inflow to the dam29. Microclimate condition was the primary factor resulting in change of aquatic microorganism diversity for similar water quality between reservoir head region and Changjiang estuary. Therefore, the study was important significance and researchers should pay more attention to focusing on microbial communities of estuary reservoir. Furthermore, compared with the effect of sampling positions, air temperature changed the microbial community structures more violently for samples were clustered into three parts (Fig. 5).
The pyroseqencing analysis showed that Temperature, MC_RR, TN and DO were key environmental parameters for prokaryotic communities and Temperature and DO were also the critical factors for eukaryotic communities (Fig. 5). While compared to prokaryotes, majority of the environmental factors for eukaryotes did not explained perfectly for community structure changes. Therefore, roots of changes were derived from the species themselves, really not constrained variables. Transparency, CODMn and TN were shown to be significant factors for prokaryote from late August to mid-October. A strong relationship was also observed among transparency, TN, DO and the eukaryotic community during winter and spring. Moreover, chlorophyll-a had a close affiliation to both prokaryotic algae in summer and autumn and eukaryotic algae in winter and spring. Transparency also had a strong relationship with algae caused by the increase in algae biomass. Nutrients, such as TN and TP, were shown to be weakly related and TN had a close relation to both prokaryotic algae in summer and autumn and eukaryotic algae in winter and spring. Data revealed that the TN and TP concentrations had significant discrepancy among the locations (S1). Thus, TN was related to the algae community throughout the year, whereas TP had a lower effect, contrary to the findings of another study conducted on Lake Taihu. TN and TP were also considered to be the major environmental factors at the hypertrophic sampling site35. CODMn represented the status of water pollution by organic compounds. This study demonstrated that high CODMn was associated with the rapid proliferation of Microcystis. The concentration of microcystin (MC-RR and MC-LR) dropped to the lowest level during Microcystis rapid proliferation, which was attributed to the intracellular microcystin that was not released into the water. Low water temperature is accompanied by high DO value and this study showed that low DO had a weaker relation with Cyanophyta bloom38. Shu3et al reported some similar results that the combined shared features of temperature and nutrition conditions led to relatively high abundances of total Microcystis and other results with minor difference that the concentrations of chlorophyll-a and microcystin were higher than those found in this study. This knowledge is valuable for in-depth understanding of microbial community biodiversity in estuary reservoirs, particularly those that are important potential water sources.
Methods
Description of sampling sites
A total of 80 samples were collected monthly from February to October 2013. The sampling region was located in the ChangjiangEstuary and samples were collected according to climate and the four seasons. Spring started on March 4 and the temperature increased from 10°C to 22°C. Summer started on May 5 and the temperature was above 22°C. Autumn started on October 15 and the temperature decreased from 22°C to 10°C. Winter started on November 28 and the temperature was below 10°C. Phytoplankton reproduced rapidly in summer. Six sampling locations were selected for environmental heterogeneity, as shown in Fig. 1. The sampling sites were as follows: (1) Site 0 was the water inlet of the reservoir; (2) Site 2 was situated south of the reclamation area; (3) Site 3 was located north of the reclamation area; (4) Site 8 was at the center of the reservoir; (5) Site 13 was near the water outlet and water intake of the reservoir; (6) Site 15 was the tail of the reservoir. The samples were evaluated by sampling time and sampling station and were marked from 0226-0 (month date-point) to 1029-13 in sequence.
Reservoir characteristics analysis
Water samples from the estuary reservoir were collected using a 500 ml water sampler (WB-PM, Beijing Pulite Instrument Co. Ltd., China). Water temperature was instantaneous value measured using a multi-parameter water quality analyzer (Multi3410, WTW Company, Germany) and air temperature was average value from local meteorological bureau. Water quality indices, including DO, CODMn, TN, TP, transparency and chlorophyll-a, were analyzed using the standard methods39. Microcystin was determined using an Agilent 1260 High Performance Liquid Chromatography (HPLC)40. Microcystin standard substances (MC-LR and MC-RR) were purchased from Sigma, guaranteed reagents were bought from Fisher, other laboratory reagents were obtained from Sinopharm. Freshwater was sampled at a depth of approximately 0.5 m from the top water layer. Microorganisms were collected by filtering 500 ml of the collected water through hydrophilic nuclepore filters (0.22 μm, Jingteng Laboratory Equipment Co. Ltd., China) and then stored at −80°C.
DNA extraction, amplification and sequencing
Genomic DNA was extracted from the same amount of membrane (0.1 g) on the day after sampling by using the E.Z.N.A. Stool DNA Kit (Omega, USA) according to the manufacturer alternative protocol previously described by Bosshard et al41. The V1-V3 region of the 16S rRNA gene42 was amplified using the forward primer 8F (5′-AGAGTTTGATCCTGGCTCAG-3′) and reverse primer 533R (5′-TTACCGCGGCTGCTGGCAC-3′). The V4 region of the 18S rRNA gene was amplified using 3NDf (5′-GGCAAGTCTGGTGCCAG-3′) and reverse primer V4_euk_R2 (5′-ACGGTATCTATCTCTTCG-3′). Polymerase chain reaction was conducted in 20 μl reaction mixtures containing 2 ul of deoxynucleoside triphosphate at a concentration of 2.5 mM and 0.8 μl of forward and reverse primers, respectively (5 μm each), 0.4 μl FastPfu Polymerase, 5 × FastPfu Buffer 4 μl and 10 ng of DNA. Each reaction mix received 1 μl of genomic community DNA as a template. The following cycling parameters were used for prokaryotes: retain 2 min at 95°C, 25 cycles (95°C for 30 s, 55°C for 30 s and 72°C for 30 s), followed by a final extension at 72°C for 5 min, until halted at 10°C by the user. Similar reaction parameters were used for eukaryotes for 35 cycles (95°C for 30 s, 55°C for 30 s and 72°C for 45 s). Triplicate reaction mixtures were pooled per sample, purified using an AxyPrep DNA gel extraction kit (Axygen, USA) and quantified using the QuantiFluor-ST Fluorescence quantitative system (Promega, USA). The mixtures were then pyrosequenced using a Roche 454 Genome Sequencer FLX Titanium platform (Majorbio Bio-Pharm Technology Co. Ltd., Shanghai, China) as previously described28,43.
Processing of pyrosequencing data
Data were processed using the Quantitative Insights Into Microbial Ecology (Qiime) pipeline for 16S rRNA and 18S rRNA data sets (http://qiime.org/tutorials/index.html)44. In brief, the sequences with a quality score below 25 and 200 bp in length were trimmed and then assigned to water samples based on unique 10-bp barcodes. Sequences were binned into operational taxonomic units (OTUs) using a 97% identity threshold and the most abundant sequence from each OTU was selected as a representative sequence for that OTU. Taxonomy was aligned and compared with the SILVA database (SILVA version 115)45. To compare richness between different treatments, Qiime randomly chose a series of subsets of each library in different sizes (100, 200, 300, 400, 500, 600... with a step of 100) to calculate respective richness indices. Bar plots were done by Microsoft Excel 2010 and heatmap analysis were conducted using the R-gplots for Linux with the clustering of the different samples. The analysis was carried out without transformation of data and focused on scaling of original data. The abline() parameter from R-graphics was used to determine whether unimodal or linear method was appropriate. Axis lengths of DCA1 of bacterial and fungal sequences were 3.20 and 3.53 respectively and these value did not indicate a clear linear or unimodal relationship. Therefore, RDA was more suitable with a short gradient. RDA using the R-vegan and R-maptools for Linux was selected to compare species-environment correlations46.
References
Arscott, D. B., Dow, C. L. & Sweeney, B. W. Landscape template of New York City's drinking-water-supply watersheds. J N Am. Benthol. Soc. 25, 867–886 (2006).
Bayley, R., Ta, C. T., Sherwin, C. J. & Renton, P. J. Traditional and novel reservoir management techniques to enhance water quality for subsequent potable water treatment. 6th International Conference of the IAWQ-IWSA Particle-Separation-Group, 15-18 Febrary 2000, Durban, South Africa. Particle Removal from Reservoirs and other Surface Waters 1, 9–16 (2001).
Shu, H. T. & Karina, Y. H. G. The dynamics of cyanobacteria and microcystin production in a tropical reservoir of Singapore. Harmful Algae 10, 319–329 (2011).
Derek, P. T. Y. & Tang, S. L. Sustainable strategies on water supply management in Hong Kong. Water. Environ. J. 25, 192–199 (2011).
Celussi, M. & Cataletto, B. Annual dynamics of bacterioplankton assemblages in the Gulf of Trieste (Northern Adriatic Sea). Gene 406, 113–123 (2007).
Torsvik, V., Daae, F. L., Sandaa, R. A. & Ovreas, L. Review article: novel techniques for analyzing microbial diversity in natural and perturbed environments. J Biotechnol. 64, 53–62 (1998).
Zelles, L. Fatty acid patterns of phospholipids and lipopolysaccharides in the characterisation of microbial communities in soil: a review. Biol. Fert Soils. 29, 111–129 (1999).
Ramsey, P. W., Rillig, M. C., Feris, K. P., Holben, W. E. & Gannon, J. E. Choice of methods for soil microbial community analysis: PLFA maximizes power compared to CLPP and PCR-based approaches. Pedobiologia 50, 275–280 (2006).
Margulies, M., Egholm, M., Altman, W. E., Attiya, S. & Bader, J. S. Genome sequencing in microfabricated high-density picolitre reactors. Nature 437, 376–380 (2005).
Glenn, T. C. Field guide to next generation DNA sequences. Mol. Ecol. Resour. 11, 759–769 (2011).
Logares, R., Sunagawa, S. & Salazar, G. Metagenomic 16S rdna illumina tags are a powerful alternative to amplicon sequencing to explore diversity and structure of microbial communities. Environ. Microbiol. 10.1111/1462-2920.1225 (2013).
Tas, S., Yilmaz, N. & Okus, E. Phytoplankton as an indicator of improving water quality in Golden Horn Estuary. Estuar. Coast. 32, 1205–1224 (2009).
Paterson, R. R. M. & Lima, N. Survey and significance of filamentous fungi from tap water. Int. J Hyg. Envir. Heal. 209, 257–264 (2009).
Kravchuk, E. S. & Anisimova, T. V. Potential effect of macrophyte beds on number of akinetes of blue-green algae in littoral of a body of water. Contemp. Probl. Ecol. 5, 430–433 (2012).
Stoeck, T. et al. Multiple marker parallel tag environmental DNA sequencing reveals a highly complex eukaryotic community in marine anoxic water. Mol.Ecol. 19, 21–31 (2010).
Kim, J. et al. Hydrodynamic effects on bacterial biofilm development in a microfluidic environment. Lab. Chip. 13, 1846–1849 (2013).
Yan, Q. Y., Yu, Y. H., Feng, W. S., Yu, Z. G. & Chen, H. T. Plankton community composition in the three gorges reservoir region revealed by PCR-DGGE and its relationships with environmental factors. J Environ. Sci. 20, 732–738 (2008).
Fonseca, B. M. & Bicudo, C. E. M. Phytoplankton seasonal variation in a shallow stratified eutrophic reservoir (Garcas Pond, Brazil). Hydrobiologia 600, 267–282 (2008).
Cheng, C. et al. Bar-coded pyrosequencing reveals the bacterial community during Microcystis water bloom in Guanting Reservoir, Beijing. Procedia. Engineering. 18, 341–346 (2011).
Znachor, P., Zapomelova, E., Rehakova, K., Nedoma, J. & Simek, K. The effect of extreme rainfall on summer succession and vertical distribution of phytoplankton in a lacustrine part of a eutrophic reservoir. Aquat. Sci. 70, 77–86 (2008).
Boucher, D., Jardillier, L. & Debroas, D. Succession of bacterial community composition over two consecutive years in two aquatic systems: a natural lake and a lake-reservoir. FEMS Microbiol. Ecol. 55, 79–97 (2006).
Edward, J. P. et al. Climatic influences on autochthonous and allochthonous phytoplankton blooms in a subtropical estuary, St. Lucie Estuary, Florida, USA. Estuar. Coast. 35, 335–352 (2012).
Seyfettin, T., Noyan, Y. & Erdogan, O. Phytoplankton as an indicator of improving water quality in the Golden Horn Estuary. Estuar. Coast. 32, 1205–1224 (2009).
Muyzer, G., Waal, E. C. D. & Uitterlinden, U. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl. Environ. Microb. 59, 695–700 (1993).
Bernhard, A. E., Donn, T., Giblin, A. E. & Stahl, D. A. Loss of diversity of ammonia-oxidizing bacteria correlated with increasing salinity in an estuary system. Environ. Microbiol. 7, 1289–1297 (2005).
Mosier, A. C. & Francis, C. A. Relative abundance and diversity of ammonia-oxidizing archaea and bacteria in the San Francisco Bay estuary. Environ. Microbiol. 10, 3002–3016 (2008).
Zeng, Y. X. et al. Bacterioplankton community structure in the Arctic waters as revealed by pyrosequencing of 16S rRNA genes. Anton. Leeuw. Int. J G. 103, 1309–1319 (2013).
Brate, J. et al. Freshwater Perkinsea and marine-freshwater colonizations revealed by pyrosequencing and phylogeny of environmental rDNA. The ISME J. 4, 1144–1153 (2010).
Pavel, R. & Petr, Z. Spatial heterogeneity and seasonal succession of phytoplankton along the longitudinal gradient in a eutrophic reservoir. Hydrobiologia 663, 175–186 (2011).
Barone, R. & Naselli-Flores, L. Distribution and seasonal dynamics of Cryptomonads in Sicilian water bodies. Hydrobiologia 502, 325–329 (2003).
Zeng, D. N. et al. Analysis of the bacterial communities associated with different drinking water treatment process. World. J Microb. Biot. 29, 1573–1584 (2013).
Langille, M. G. I. et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–823 (2013).
Klausen, C. et al. Abundance of actinobacteria and production of seosmin and 2-methylisoborneol in Danish streams and fish ponds. FEMS Microbiol. Ecol. 52, 265–278 (2005).
Kasalicky, V., Jezbera, J., Hahn, M. W. & Simek, K. The diversity of the Limnohabitans Genus, an important group of freshwater Bacterioplankton, by Characterization of 35 isolated strains. PLoS ONE 8, e58209 (2013).
Tian, C. et al. Spatiotemporal transition of bacteriaplankton diversity in a large shallow hypertrophic freshwater lake, as determined by denaturing gradient gel electrophoresis. J Plankton. Res. 31, 885–897 (2009).
Te, S. H. & Gin, K. Y. H. The dynamics of cyanobacteria and microcystin production in a tropical reservoir of Singapore. Harmful Algae 10, 319–329 (2011).
Feng, B. W. et al. Bacterial diversity of water and sediment in the Changjiang estuary and coastal area of the East China Sea. FEMS Microbiol. Ecol. 70, 236–248 (2009).
Havens, K. E. Cyanobacteria blooms: effects on aquatic ecosystems. Adv. Exp. Med. Biol. 619, 733–747 (2008).
State Environmental Protection Administration. Water and wastewater monitoring analysis method of editorial committee: Water and wastewater monitoring analysis method, 4rd (China Environmental Science Press, Beijing, 2002).
Murata, H. et al. High performance liquid chromatography with chemiluminescence detection of derivatized microcystins. J Chromatogr. A 693, 263–270 (1995).
Bosshard, P. P., Santini, Y., Gruter, D., Stettler, R. & Bachofen, R. Bacterial diversity and community composition in the chemocline of the meromictic alpine Lake Cadagno as revealed by 16S rRNA gene analysis. FEMS Microbiol. Ecol. 31, 173–182 (2000).
Huse, S. M. et al. Exploring microbial diversity and taxonomy using SSU rRNA hypervariable tag sequencing. PLos. Genet. 4, e1000255 (2008).
Wu, S. et al. Composition, diversity and origin of the bacterial community in grass carp intestine. PLoS ONE 7, 1–10 (2012).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods. 7, 335–336 (2010).
Pruesse, E. et al. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids. Res. 35, 7188–7196 (2007).
Legendre, P., Oksanen, J. & Braak, C. F. J. Testing the significance of canonical axes in redundancy analysis. Methods Ecol. Evol. 2, 269–227 (2011).
Acknowledgements
We thank Shanghai Chengtou Raw Water Company Limited for their complete financial support for this project, as well as for the help of water sample collection and some advices in manuscript. We also thank Prof. Haizheng Li, Tongji University for his useful suggests. We are expressing our many thanks for Satellite Environment Center, Ministry of Environmental Protection in China [http://www.secmep.cn/secPortal/portal/column/itemDetails.faces?itemid=8ae67f88272ce22801272d0f50410005] which supplies their free availability of HJ-1 satellite data with license of data usage in Chinese. Moreover, we express our thanks for Dr. Hao Zhang, Department of Environmental Science and Engineering, Fudan University, who synthesized the image of Figure 1 using those free HJ-1 satellite data by commercial software of GeoStar 3.0 (http://www.geostar.com.cn/index.php?m=content&c=index&a=lists&catid=84), as well as thanks for Free Software Foundation (FSF) (http://www.fsf.org) which supports GNU Image Manipulation Program (http://www.gnu.org) supplying Gimp 2.8.10 software in free of charge (http://www.gimp.org) for our modification to Figure 1.
Author information
Authors and Affiliations
Contributions
This work was conceived by Y.L. laboratory work was done by Z.S. and C.W.W. and Y.H.J. Samples were provided by G.P.L. and Y.P.Z. and S.M.Z. Analysis was carried out by Z.S. and Y.L. and G.P.L. The manuscript was written and approved by Z.S. and Y.L.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplemental Dataset S1
Supplementary Information
Supplementary Files 2 & 3
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Sun, Z., Li, G., Wang, C. et al. Community dynamics of prokaryotic and eukaryotic microbes in an estuary reservoir. Sci Rep 4, 6966 (2014). https://doi.org/10.1038/srep06966
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep06966
This article is cited by
-
Utility of investigation for suspected microbial keratitis: a diagnostic accuracy study
Eye (2023)
-
Microbial Communities Are Shaped by Different Ecological Processes in Subtropical Reservoirs of Different Trophic States
Microbial Ecology (2023)
-
Seasonal co-occurrence patterns of bacteria and eukaryotic phytoplankton and the ecological response in urban aquatic ecosystem
Journal of Oceanology and Limnology (2022)
-
The effect of graphene photocatalysis on microbial communities in Lake Xingyun, southwestern China
Environmental Science and Pollution Research (2022)
-
Seasonal and spatial patterns of eukaryotic phytoplankton communities in an urban river based on marker gene
Scientific Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
