
Abstract
Chaperonins mediate protein folding in a cavity formed by multisubunit rings. The human CCT has eight non-identical subunits and the His147Arg mutation in one subunit, CCT5, causes neuropathy. Knowledge is scarce on the impact of this and other mutations upon the chaperone's structure and functions. To make progress, experimental models must be developed. We used an archaeal mutant homolog and demonstrated that the His147Arg mutant has impaired oligomeric assembly, ATPase activity and defective protein homeostasis functions. These results establish for the first time that a human chaperonin gene defect can be reproduced and studied at the molecular level with an archaeal homolog. The major advantage of the system, consisting of rings with eight identical subunits, is that it amplifies the effects of a mutation as compared with the human counterpart, in which just one subunit per ring is defective. Therefore, the slight deficit of a non-lethal mutation can be detected and characterized.
Similar content being viewed by others


Introduction
Molecular chaperones are indispensable cellular components that assist folding and assembly of newly synthesized proteins, translocation of proteins across membranes, as well as refolding and degrading of misfolded and aggregated proteins1. A class of chaperones, the chaperonins (Cpns) are large, hollow, ATP-dependent nanomachines that promote correct folding of a wide range of proteins2,3,4,5,6,7. Cpns are divided into two groups: group I Cpns, represented by GroEL/GroES, in bacteria, mitochondria and chloroplasts and group II Cpns, occurring in eukaryotes and archaea8,9,10. They share similar quaternary structures consisting of a double toroid cylinder assembled into two rings stacked back to back. This arrangement generates two dynamic internal cavities that allow unfolded proteins to fold correctly in a confined environment that opens and shuts in a cycle powered by ATP hydrolysis11,12. Group I Cpns are composed of 14 identical subunits arranged in two heptameric rings and require a co-chaperone, GroES, to facilitate protein folding. The Group II Cpns, which are very similar in Archaea and Eukarya, form double rings with an eight or nine fold rotary symmetry and have built-in lids to function without a co-chaperone13,14,15.
The human CCT (chaperonin containing TCP1) is a complex assembly of eight similar but nonidentical subunits which functions to fold non-native proteins through the alternative opening and closing of the two chambers. By contrast, many hyperthermophilic Archaea, including Pyrocococcusfuriosus and several groups such as Pyrodictium, Methanopyrus, and Pyrobaculum spp have a single group II Cpn subunit. The double ring of these archaea is therefore composed of 16 identical subunits and is minimally complex16. Hyperthermophile Cpns are exceptionally stable in vitro17, affording a suitable model for studying oligomerization of group II Cpns such as the human CCT complex.
Mutations in human Group II CCT subunits 4 and 5 cause rare debilitating diseases18,19,20. For example, the His147Arg mutation in human CCT5 causes mutilating peripheral sensory neuropathy20. Little is known concerning the pathogenic mechanisms of these CCT mutations at the molecular level and on the impact of the mutations upon the structure and function of the complex. Mutations that allow survival of the carriers are likely to have very subtle mechanistic deficits since major functional disruption would be lethal due to the critical and multiple roles of this ubiquitous chaperone. Therefore, pathogenic mutations with living phenotypes have to be considered a priori difficult to characterize at the molecular level with the systems available today. Ways must be found to amplify a defect to make it measurable. For this purpose, we focused on modeling the CCT5 mutation, using the Cpn60 from Pyrococcus furiosus (Pf), which has only one subunit homolog that assembles into a hexadecameric double-ring complex like the human counterpart21.
Results
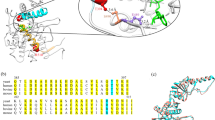
Our strategy to use the CCT homolog from a hyperthermophile was based on the conservation of the sequence and inferred structure of the human CCT5 and the archaeal subunit shown in Fig. 1 (see also Supplementary Fig. S1). The colinearity of the alignment and the high similarity of the sequences suggest that the double-ring structure would be a simplified version (homo-oligomer) with functional equivalence to the extremely complex (hetero-oligomer) human CCT hexadecamers. The archaeon Pyrococcus furiosus grows optimally at 100°C. In a previous study we have characterized a C-terminal structural motif that is a major determinant of extreme thermal stability. Deletion or modification of this sequence does not disrupt the hexadecameric structure, but “detunes” the activity without loss of ATPase or protein folding properties22. The deletion of 22 amino acids from the C-terminus of the Pf Cpn60 removes the “modulator domain” that controls thermostability in this exceptionally thermostable protein. Consequently, the thermal stability and Tmax for activity are downgraded to 50°C. In Fig. 1, the alignment of the archaeal Cpn60 (PfCD) and the human CCT5 shows that there is strong conservation between these polypeptides. The human CCT5 has 44% amino acid sequence identity and 65% similarity with PfCD. The amino acid His147 in CCT5 matches Ile138 in PfCD and the upstream and downstream sequences show conservation and colinearity.

The N-terminal portion of the amino-acid sequence alignment of human CCT5 (uniprot ID; P48643) and Pyrococcus furiosus PfCPN (uniprot ID; Q8TZL6) (A) with a 22-amino acids deletion in C-terminus, named PfCD (see B).
For a complete alignment see Supplementary Fig. S1. The residues conserved between human CCT5 and PfCD are shown in the space between their sequences. The secondary structures were predicted using the GOR IV server (http://npsa-pbil.ibcp.fr/cgi-bin/npsa_automat.pl?page=npsa_gor4.html) and α-helix, β-sheet and loop elements are represented as red rings, blue wave and black line, respectively, above or below their amino-acid sequences. The amino acids matching the human pathogenic mutation in CCT5 (His147) and the equivalent locus in the archaeal PfCD (Ile138) that we mutated to Ile138His and Ile138Arg - the humanized archaeal molecules - are shown in red bold letters and indicated with an asterisk.
Structural representations of the human CCT5 and PfCD were generated in the Swiss-Model server, using their primary amino-acid sequences and as template the crystal structure of the Cpn60 α subunit from Thermococcus KS-1 (KS-1α; PDB ID: 1Q3Q), which was crystallized with an ATP analogue, AMP-PNP and fluoride ion in the active site23. Overall structural similarities and characteristic features of group II chaperonins are evident around the equatorial domain (Fig. 2A). This domain comprises the ATP/ADP binding site, is the region where the mutation H147R occurs in the case of the human CCT5-associated neuropathy and is essentially superimposed. Noteworthy, the α-helixes in which are located His147 in CCT5, or Ile138 in PfCD overlap and the side chain of His147 of human CCT5 and Ile138 of PfCD showed the same configuration as Ile138 of KS-1α. To analyze the structural consequences of mutation I138R in PfCD, we superimposed the PfCD structure onto the crystal structure of the KS-1α (1Q3Q) (Fig. 2B). The mutation I138R is located in the equatorial domain, in the N-terminus of Cpn and this position is close to the intermediate domain, which serves as a hinge to allow binding of ATP/ADP when the apical alpha-helical protrusion domain of Cpn recognizes client proteins24. Arg138 is contiguous to the conserved motif Gly94-Thr99 constituting the ATP binding site in group II chaperonins25, suggesting that steric hindrance might impede the conformational change necessary for ATP binding. A similar situation is apparent in the case of mutant human CCT5 with Arg147, instead of His147 (Fig. 2C).

(A). Human CCT5 and P. furiosus PfCD superposed onto the crystal structure of Thermococcus strain KS-1 α. LEFT: The structure (Swiss-Model (http://swissmodel.expasy.org/) of PfCD (gold) superposed onto the crystal structure of KS1α (momomers, displayed in marine blue, violet and deep teal as surface and in cyan color as ribbon). The hexadecamer, double-ring structure, is depicted by a dotted line, as is on the right of the subsequent panels B and C. RIGHT: Magnified image of the superposed structures of the PfCD (gold) and Human CCT5 (orange) onto KS1α; cyan ribbon). Side chains of isoleucine at 138 of PfCD (blue) and isoleucine at 138 of KS1α (deep teal) and side chain of histidine at 147 of human CCT5 (red) are represented as ball and stick. AMP-PNP (green stick; also shown on the left in panels B and C) and magnesium ion (yellow ball). (B). PfCD around the Arg138 mutation and its predicted matching areas in the crystal structure of KS1α. LEFT: Mutant Arg138 (orange) and its adjacent Arg421 (red) in the opposite α helix are drawn with stick in the PfCD structure (marine blue). RIGHT: Arg138 mutation area was superposed onto the crystal structure of KS1α (monomers are displayed in various shades of blue). (C). Human CCT5 structure around the Arg147 mutation viewed from different angles and predicted matching areas in the crystal structure of KS1α. LEFT: Wild-type His147 (blue), mutant Arg147 (red) and their adjacent amino acids, Ser428 and Cys429 (magenta) in the opposite α helix are drawn with ball and stick and transparent surface in the human CCT5 structure (orange ribbon). RIGHT: The predicted Arg147 mutation area in the monomers is shown as a red surface on the crystal structure of KS1α (monomers are shown as surface in marine blue, violet, deep teal and cyan colors.
In Fig. 3, the optimal temperatures for ATPase activity of the Pf Cpn wild type as well as 1138H and I138R were shown to be 50°C. The PfCD and I138H Cpns showed much higher ATPase activities than I138R Cpn over the whole temperature range investigated. Fig. 3 also shows that the residual activity of I138R Cpn was ~12% of that of PfCD, whereas I138H retained ~88% of ATPase activity compared with PfCD. These data indicate that Arg located at position 138 caused the loss of ~88% of the total ATPase activity displayed by PfCD.

ATPase activities of PfCD, I138H and I138R Cpns at various temperatures.
PfCD (red square), I138H (green triangle), I138R (purple star). The results shown are mean values (±SD) of triplicate experiments.
Fig. 4 shows the wild type, I138H and I138R complexes separated on a 4–9% gradient native-PAGE gel. Group II Cpns typically form double-ring hexadecamers, the quaternary structures of group II Cpn, consisting of a double toroid cylinder assembled into two rings of subunits stacked back-to-back. This conformation is optimal for promoting correct folding of non-native cellular proteins22. Whereas there was no clear difference in molar concentration ratios of hexadecamer between PfCD and I138H, a relatively lower concentration of double-ringed Cpn was observed in the case of I138R (Fig. 4, top panel). Densitometric analysis revealed that I138 formed double ring approximately 4–6 fold less than PfCD or I138H, indicating a functional deficit in maintaining the double ring structure (results not shown).

Oligomeric states of purified PfCD, I138H and I138R Cpns determined by gel filtration chromatography (top panel) and corresponding SDS-PAGE analysis (bottom panel).
PfCD, or I138H, or I138R purified by gel filtration chromatography followed by anion exchange chromatography were pooled together for ATPase activity and MDH heat-protection activity assays. The same amounts of these pooled pure Cpn proteins were subjected to 4–9% gradient native-PAGE analysis (top panel) and 12% SDS-PAGE analysis (bottom panel) and then stained with Coomassie brilliant blue G-250. The figures to the right, 16 on top and 1 at the bottom, indicate hexadecamer and monomer, respectively, while the other oligomers are distributed in between them as shown by the gel bands. The thick arrow at the very bottom indicates the monomeric band of the purified Cpns run with same amount of protein in SDS-PAGE. St, molecular weight standard.
Fig. 5 shows that protection against heat denaturation of two enzymes by PfCD and its mutants is lower for I138R as compared with I138H and PfCD when hetero-oligomeric preparations are tested. However, hexadecamers of all three variants of the chaperonin, freshly purified by gel permeation chromatography, all showed intact heat protection activity. The enzymes tested at various temperatures were porcine mitochondrial malate dehydrogenase (MDH) and shrimp alkaline phosphatase (SAP). The results with hetero-oligomeric preparations, incubated with MDH at 37 or 42°C are shown in panels A and B, respectively, while the results with freshly purified hexadecamers are shown in panels C (MDH, at 37°C) and D (SAP, at 50°C).

MDH heat-protection activity profiles of mixed oligomers of PfCD, I138H and I138R Cpns at 37°C (A) and 42°C (B); and MDH heat-protection activity profiles at 37°C (C) and SAP heat-protection activity profiles at 50°C (D) of pure hexadecameric Cpns.
MDH (or SAP, in panel D) residual activities in the presence of PfCD (red square), I138H (green triangle), or I138R (purple star). Negative control, MDH (or SAP in panel C) residual activity without any Cpn (blue diamond). The results shown are mean values (±SD) of triplicate experiments.
In Fig. 6, the ability of the archaeal Cpn60 to deconstruct and disperse amyloid fibrils under mild conditions, a system that has been described in detail elsewhere26, is confirmed. The top panel shows the activity of the PfCD Cpn60 to disrupt fibrils completely in 30 min. The activity of the I138H mutant is similar to PfCD (middle panel), whereas the I138R mutant is defective and the fibrils are intact at 60 min (bottom panel).

Dispersion of amyloid fibrils by archaeal PfCD (top strip of panels), partial dispersion by I138H (middle strip of panels) and lack of dispersion by I138R (bottom strip of panels).
Since the study of the activity of pure monomers was not feasible because of their strong tendency to oligomerize, their response to heat stress was examined in silico, via simulations. The results are shown in Fig. 7 and the overall conclusions from these analyses are displayed in Table 1. It can be seen that the archaeal chaperonin with optimal temperature at 42°C, or higher, is rigid (it loses flexibility) at 37°C, a stressful temperature for this molecule. Similarly, CCT5 is flexible at its physiological temperature of 37°C but is rigid, i.e., it loses flexibility, at the stressful temperature of 42°C. It can also be seen that the Arg mutations in both the archaeal and the human chaperonins, result in a loss of flexibility in the two molecules, even at their respective optimal temperatures.

Root mean square deviation (RMSD) plot of Cα atoms at 310 and 315 K. Top panel: PfCD at 310 K (blue line) and at 315 K (red line).Middle panel, I138H at 310 K (blue line) and at 315 K (red line); I138R at 310 K (green line) and at 315 K (violet line). Bottom panel: Human CCT5 with H at position 147 at 310 and 315 K (blue and red lines, respectively); Human CCT5 with R at position 147 at 310 and 315 K (green and violet lines, respectively).
Discussion
Human TRiC/CCT is a double-ring complex that interacts with ~10% of the human cytosolic proteins and is essential to achieve folding and stabilization of many essential proteins in human27 and yeast28. In addition, TRiC modulates and suppresses the aggregation of neurologically toxic proteins with polyglutamine motifs29,30. We have set out to model the function of mutant forms of the TRiC hexadecamer using an archaeal Cpn60 homolog with high sequence and structural homology to CCT5 (Fig 1). As shown in Fig. 2, when the modeled CCT5 structure was superimposed onto the crystal structure of the Cpn60 subunit from the archaeal Cpn60, they showed overall structural similarities and characteristic features of group II chaperonins around the equatorial domain (Fig. 2A), including the ATP/ADP binding site, which is the region where the mutation His147Arg occurs in the case of the human CCT5-associated neuropathy. Remarkably, the α-helixes that include His147 in CCT5 and Ile138 in PfCD overlap and the side chain of His147 of human CCT5 and Ile138 of the archaeal model colocated with Ile138. Assuming that His 147 was the wild type residue at the position of the mutation causing the peripheral sensory neuropathy, we mutated Ile138 to His to recreate the human structural element and tested the effect on chaperone stability and function in vitro. H138R is located in the equatorial domain, near the N-terminus, close to the intermediate domain, which serves as a hinge with a large articulation angle, to allow binding of ATP/ADP when the apical alpha-helical protrusion domain of Cpn recognizes client proteins24. Interestingly, our I138R structure representation revealed that the Arg138 mutation formed a close contact with a neighboring Arg421 in the adjacent α-helix. It seems that the side chain of Arg could cause a steric hindrance through the repulsive effects of closing the distance between two internal positive charges, thus affecting the helix-helix interface (Fig. 2C). Arg138 is contiguous to the highly conserved motif Gly94-Thr99 for ATP binding in group II chaperonins25, suggesting steric hindrance might impede the necessary conformational change for ATP binding.
Figure 2 also shows that the “humanized” version had little effect on the structure and function of the complex. In contrast, the mutation I138R (Fig. 2C), sharply reduced the catalytic capability resulting in low ATPase activity and poor heat protection of mesophilic vertebrate enzymes. The quaternary structures of group II chaperonins, consisting of two rings of subunits stacked back to back, were determined on a gradient native-PAGE gel shown in Fig. 4, similar to analysis of assembly deficient N terminal mutants22. Interestingly, whereas there was no difference in molar concentration ratios of hexadecamer in preparations of PfCD, or I138H, double-ringed Cpn at ~1 mDa was sharply reduced in the case of I138R (Fig. 4, top panel). Densitometric analysis revealed that I138R formed less than 20% of double rings than PfCD or I138H indicating a functional deficit in maintaining the double ring structure (Supplementary material).
We have previously characterized the C-terminus as a primary thermostability determinant, which can be tuned to achieve lower temperatures for optimal activity of the archaeal Cpn6022. Thus, the Cpn60 complex from the hyperthermophile Pyrococcus furiosus (Topt for growth 100°C), which was originally active and stable at 95°C, was lowered to 50–55°C by the deletion of the C-terminus. Thus detuned, the Cpn60 can be used for refolding and salvage of mesophilic enzymes such as porcine mitochondrial MDH as shown in Fig. 5. Porcine MDH is known to lose activity rapidly at 51°C31. Incubation at 50°C in the absence of Cpn resulted in rapid first-order loss of MDH activity (t1/2: 13 min). Both the PfCD and I138H Cpns were effective in protecting MDH against heat-denaturation at both 42 and 50°C compared to unprotected MDH (Figs 5A and B, respectively). By contrast, I138R showed drastically diminished inhibition of heat-denaturation of MDH (t1/2: 36 min), as indicated by ~8.9-fold decrease in the half-life (t1/2) compared to that of MDH protected by PfCD and I138H. At 42°C a similar trend was observed: PfCD: t1/2: 17.05 min, I138H t1/2: 15.05 min, I138R t1/2: 5 min, no Cpn t1/2: 2.7 min). Similar results were obtained with SAP at 50°C (Fig. 5D). We conclude that defective oligomerization leads to impaired chaperoning functions of the archaeal Cpn60 and that the human TRiC complex would likely display a similar effect.
Recent work has implicated TRiC as a critical defense strategy forestalling the formation of amyloid aggregates of proteins that cause devastating neurological deficits in Parkinson's and Huntington's syndromes29,30. Our group has recently constructed archaeal Cpn60 mutants to model the disaggregation and dispersal of amyloid fibrils under physiological conditions32. In Fig. 6 we show the rapid dispersal of amyloid fibrils by both the PfCD and His138 mutant chaperonin oligomers, as we have reported previously. The Arg138 mutant was completely inactive in this assay, which reflects the ability of the chaperonins to deconstruct extremely stable cross-beta structures. In past studies we have shown that highly stable and toxic prion fibrils can also be dispersed in a similar assay33. Fig. 7 confirms that the presence of Arg in place of His leads to a loss of flexure and we speculate that this results in decreased induced fit interaction during assembly.
The findings from MD simulation summarized in Table 1 are that stress, i.e., 37°C for PfCpn and 42°C for CCT5, causes loss of flexibility and the same effect is caused by the His-to-Arg mutation, which is apparent at the optimal temperatures for the two molecules, i.e., 37°C for CCT5 and 42°C for PfCpn.
It can be assumed that the devastating phenotypic effects of this mutation, which required peripheral amputation in some homozygous individuals, actually results from a very subtle biochemical defect due to pivotal role of TRiC in the posttranslational maturation of many essential proteins. Here, we demonstrate that a homooligomeric archaeal model Cpn60 complex, adjusted to fit the human temperature range, effectively amplifies this slight defect eight-fold compared to the human TRiC complex, composed of eight nonidentical subunits per ring.
Methods
Sequence comparison
The following molecules were studied and compared: Human CCT5 (CCT5) wild type (His147) and its mutant CCT5 Arg147 (His147Arg); Pyrococcus furiosus Chaperonin 60 with Ile138, with the last 22 amino acids deleted (PfCD)17,34; wild type for this work); PfCD mutant Ile138His and PfCD Arg138 (His138Arg; equivalent to the human His147Arg mutant).
Homology modelling
The 3D structures of the wild type molecules (PfCD and CCT5-His147) and their mutants (PfCD Ile138His and His138Arg; and CCT5 Arg147) were constructed by homology modeling. This was performed using PRIME software (PRIME, 2012) and FASTA sequences35. The sequences of CCT5 and PfCD were retrieved from the Uniprot KB/TrEMBL database35. Basic Local Alignment Search Tool (BLAST) searches predicted that the crystal structure of the chaperonin from Thermococcus strain KS-1, PDB code: 1Q3R; resolution-2.9 Å36, as the most suitable average structure. Alignments between the template and the target sequences were performed using ClustalW 2.0 with default parameters37. The structures of all proteins were generated using the PRIME module (PRIME, version 3.1, Schrödinger, LLC, New York, NY, 2012). The generated models were relaxed by molecular dynamic simulations and were further validated.
Chemicals, enzymes and reagents
Restriction endonucleases, Taq polymerase and DNA ligase were purchased from New England Biolabs (Beverley, MA). Malate, ATP, EDTA, DTT, β-NADH and NAD were purchased from Sigma–Aldrich (St. Louis, MO). Porcine heart malate dehydrogenase (MDH) was purchased from Amresco (Solon, OH). PageRuler Broad Range unstained protein ladder in SDS-PAGE and NativeMark™ unstained molecular weight protein standard were from Thermo scientific (Rackford, IL) and Invitrogen (Carlsbad, CA), respectively.
Cloning, expression and purification of PfCD, I138H and I138R
The PfCD gene was amplified from Pyrococcus furiosus genomic DNA using primers containing at 5′-PfCD primer with NcoI recognition sequence and 3′-PfCD primer with BamHI recognition sequence listed in Supplementary Table S1 and then this PCR product was purified using GeneJET PCR purification kit (Thermo scientific) for digestion with NcoI and BamHI enzymes. The genes encoding PfCD, I138H or I138R were cloned initially in Escherichia coli DH5α. The pET33b(+) expression vector (Novagen, Madison, WI) was used for recombinant expression in Escherichia coli Rosetta(DE3). The cell pellets were re-suspended in exchange binding buffer (25 mM Hepes–KOH, pH 7.5) fat 25% w/v prior to cell disruption using a French Press. The soluble fraction was obtained from cell homogenate, clarified by centrifugation at 4°C prior to anion exchange chromatography and gel filtration chromatography (GFC). Elution with a 0.2–1M NaCl gradient was used. Peak fractions were purified by gel permeation using HiPrep Sephacryl S-300 HR column (3 × 55 cm) at a flow rate 1.5 ml/min. To verify homogeneity fractions were run on 12% SDS-PAGE. Protein concentration was carried out using Amicon® Ultra centrifugal Filter-100,000 MWCO (Merck Millipore, Darmstadt, Germany). Protein concentration was determined by the Bradford method (Bio-Rad Protein, Hercules, CA). See Supplementary Figs. S2-S5.
Gradient native-PAGE
For oligomeric pattern analysis of recombinant PfCD, I138H and I138R, the GFC purified samples were analyzed in 4 ~ 9% gradient native-PAGE. Stacking and two separating gel buffers with 4 or 9% acrylamide were prepared without SDS. In preparation of separating gel buffers with 9% acrylamide, glycerol plus Bromophenol Blue (0.001%) was added to a final concentration of 15% and vertical acrylamide gradient formed via specific gravity difference between 4 and 9% separating gel during gel solidification. Bromophenol Blue in separating gel buffers with 9% acrylamide served to visualize the gradient after gel formation. Purified chaperonins were preheated under ATPase activity assay conditions at 50°C for 30 min to oligomerize complexes, prior to mixing with PAGE gel sample buffer without β-mercaptoethanol and SDS. Gradient native gels were run at constant voltage (20 mV) for 3 h at 23°C and PfCD, I138H and I138R protein bands on native SDS-PAGE gel were visualized by staining with Coomassie Brilliant Blue R-250. See Supplementary Figs. S4 and S5.
ATPase activity and heat protection assays using MDH or SAP
The standard ATPase activity assay reaction buffer composed of 25 mM Hepes–KOH at pH 7.2, 300 mM KCl, 1 mM MgCl2 in the presence or absence of 0.05 mg/ml final concentration of purified PfCD, I138H or I138R protein in a final volume of 90 µl. The reaction mixture was preincubated at series of temperatures ranging from 30 to 90°C for 3 min before adding ATP (50 µM) to initiate ATP hydrolysis, as reported previously22. The reaction proceeded for 15 min at each temperature in a PCR thermocycler (Bio-Rad), followed by perchloric acid addition to a final concentration of 2% v/v to quench the reaction. The liberated Pi was determined by the Malachite Green assay at 630 nm. All reactions and controls were performed under light mineral oil (Sigma-Aldrich) for avoiding evaporation during reaction. Standard deviation was calculated from assays repeated three times.
Porcine malate dehydrogenase (MDH) heat protection by PfCD, I138H and I138R was analyzed by comparing protection activities at 37 or 42°C. MDH samples (1 mg/ml) were incubated in reaction buffer (25 mM Hepes-KOH, pH 8.0, 300 mM KCl, 1 mM MgCl2 and 0.5 M ammonium sulfate) plus 4 mM ATP at each temperature in the presence or absence of Cpn (0.2 mg/ml). Residual MDH enzyme activities after different incubation time intervals (0, 3, 6, 9, 12 and 15 min at 37°C and 0, 1, 2, 3, 4 and 5 min at 42°C) were measured immediately as described39 at 25°C in modified assay mixture (90 mM Hepes–KOH, pH 8.0, 0.22 mM â-NADH, 0.55 mM oxaloacetate (Sigma-Aldrich)). The time dependent oxidation of β-NADH was measured by recording the decreasing OD 340 nm. To measure residual shrimp alkaline phosphatase (SAP) enzyme activity with heat treatment at 50°C, we used ρ-nitrophenyl phosphate (pNPP) supported by pNPP Phosphatase Assay Kit (BioAssay Systems, Hayward, CA). The pNPP is a chromogenic substrate which was converted via SAP to ñ-nitrophenol was measured at 405 nm under alkaline conditions.
Fibril dispersion assays and Atomic Force Microscopy
Amyloid fibrils prepared from bovine insulin (I5500, Sigma-Aldrich) were subjected to dispersion assays by the chaperonins26,32. Insulin was preincubated at 70°C and pH 2.4 at a concentration of 60 mg/mL for 24 h to prepare mature fibrils. The fibrillation process was terminated by bringing the sample to 23°C and centrifuging twice at 14,000 g to remove the unfibrillated fraction. The gelatinous phase containing mature fibrils was then resuspended in pH 2.4 in HCl and used for dispersion assays.
Samples for fibril dispersion assays by chaperonins were prepared by mixing chaperonins and fibrils in a ~1:20 protein molar ratio in the presence of 2 mM ATP, 1 mM Mg2+ and 20 mM sodium acetate buffer pH 6.0. The reaction mixtures were incubated at 50°C for 60 min, with aliquots taken at 10, 30 and 60 min. In the control reaction, the chaperonin was replaced by an equal volume of the buffer at 50°C for 60 min followed by imaging.
Samples for Atomic Force Microscopy (AFM) imaging were prepared by diluting the 10-, 30- and 60-minute aliquots in a 1:100 volumetric ratio by 20 mM sodium acetate buffer pH 6.0, without ATP or Mg2+ and depositing on freshly cleaved mica discs (TED PELLA, Inc., Redding, CA) for 2 min, followed by washing with buffer to remove unbound materials, blotting and drying under flowing air. The samples were then imaged in tapping mode using a SmartSPM™-1000 atomic force microscope from AIST-NT, Inc. (Novato, CA) and Aspire CT-300 silicon AFM probes with aluminum reflex coating with a spring constant of 40 N/m and resonance frequency of 300 kHz. Images were processed using the AIST-NT SPM Control Software v. 3.3.78 (AIST-NT, Inc.).
Molecular Dynamics Simulation
The minimum energy conformers of proteins obtained from comparative modelling were used as the starting structures for MD simulation. Proteins and water molecules were used as the components for the simulation. To remove bad contacts of the modelled receptors and to achieve good starting structures, the models were refined using MDS of 5000 ps with the DESMOND package (Desmond Molecular Dynamics System, version 3.1, D. E. Shaw Research, New York, NY, 2012; Maestro-Desmond Interoperability Tools, version 3.1, Schrödinger, New York, NY. 2012)38,39. The initial structures were placed in an orthorhombic box. The TIP4W water model was used to create the aqueous environment for all models. Periodic boundary conditions were applied and the systems were further neutralized by adding appropriate counter ions (Na+).
References
Kim, Y. E., Hipp, M. S., Bracher, A., Hayer-Hartl, M. & Hartl, F. U. Molecular chaperone functions in protein folding and proteostasis. Annu. Rev. Biochem. 82, 323–355 (2013).
Kubota, H., Hynes, G. & Willison, K. The chaperonin containing t-complex polypeptide 1 (TCP-1). Multisubunit machinery assisting in protein folding and assembly in the eukaryotic cytosol. Eur. J. Biochem. 230, 3–16 (1995).
Gómez-Puertas, P., Martín-Benito, J., Carrascosa, J. L., Willison, K. R. & Valpuesta, J. M. The substrate recognition mechanisms in chaperonins. J. Mol. Recognit. 17, 85–94 (2004).
Nakagawa, A. et al. Dissection of the ATP-dependent conformational change cycle of a group II chaperonin. J. Mol. Biol. 426, 447–459, 10.1016/j.jmb.2013.09.034 (2014).
Fei, X., Yang, D., LaRonde-LeBlanc, N. & Lorimer, G. H. Crystal structure of a GroEL-ADP complex in the relaxed allosteric state at 2.7 Å resolution. Proc. Nat. Acad. Sci. USA. 110, E2958-6, 10.1073/pnas.1311996110 (2013).
Yang, D., Ye, X., & Lorimer, G. H. Symmetric GroEL:GroES2 complexes are the protein-folding functional form of the chaperonin nanomachine. Proc. Natl. Acad. Sci. U S A. 12, 110:E4298-305,. 10.1073/pnas.1318862110 (2013).
Ye, X. & Lorimer, G. H. Substrate protein switches GroE chaperonins from asymmetric to symmetric cycling by catalyzing nucleotide exchange. Proc. Natl. Acad. Sci. USA 110, E4289-97, 10.1073/pnas.1317702110 (2013).
Macario, A. J. L., Malz, M. & Conway de Macario, E. Evolution of assisted protein folding: The distribution of the main chaperoning systems within the phylogenetic domain Archaea. Front. Biosci. 9, 1318–1332 (2004).
Large, A. T. & Lund, P. A. Archaeal chaperonins. Front. Biosci. (Landmark Ed). 14, 1304–1324 (2009).
Mukherjee, K., Conway de Macario, E., Macario, A. J. L. & Brocchieri, L. Chaperonin genes on the rise: new divergent classes and intense duplication in human and other vertebrate genomes. BMC Evol. Biol. 10,64 (2010).
Yébenes, H., Mesa, P., Muñoz, I. G., Montoya, G. & Valpuesta, J. M. Chaperonins: two rings for folding. Trends Biochem. Sci. 36, 424–432 (2011).
Yamamoto, Y. Y. et al. Inter-ring communication is dispensable in the reaction cycle of Group II chaperonins. J. Mol. Biol. 426, 2667–2678, 10.1016/j.jmb.2014.05.013 (2014).
Iizuka, R. et al. ATP binding is critical for the conformational change from an open to closed state in archaeal group II chaperonin. J. Biol. Chem. 278, 44959–44965 (2003).
Douglas, N. R. et al. Dual action of ATP hydrolysis couples lid closure to substrate release into the group II chaperonin chamber. Cell. 2011 Jan 21; 144, 240–252, 10.1016/j.cell.2010.12.017 (2011).
Zhang, J. et al. Cryo-EM structure of a group II chaperonin in the prehydrolysis ATP-bound state leading to lid closure. Structure. 2011 May 11; 19, 633–639, 10.1016/j.str.2011.03.005 (2011).
Laksanalamai, P., Whitehead, T. A. & Robb, F. T. Minimal protein-folding systems in hyperthermophilic archaea. Nature Rev. Microbiol. 2, 315–324 (2004).
Luo, H., Laksanalamai, P. & Robb, F. T. An exceptionally stable Group II chaperonin from the hyperthermophile Pyrococcus furiosus. Arch. Biochem. Biophys. 486, 12–18 (2009).
Macario, A. J. L. & Conway de Macario, E. Sick chaperones, cellular stress and disease. New Eng. J. Med. 353, 1489–1501 (2005).
Macario, A. J. L., Conway de Macario, E. & Cappello, F. The Chaperonopathies. Diseases with Defective Molecular Chaperones. (eBook); 10.1007/978-94-007-4667-1 (2013). Springer Dordrecht-Heidelberg-New York-London. (2013).
Bouhouche, A., Benomar, A., Bouslam, N., Chkili, T. & Yahyaoui, M. Mutation in the epsilon subunit of the cytosolic chaperonin-containing t-complex peptide-1 (Cct5) gene causes autosomal recessive mutilating sensory neuropathy with spastic paraplegia. J. Med. Genet. 43, 441–443 (2006).
Luo, H. & Robb, F. T. Protein folding systems in thermophiles. [Chapter 4.9 pp. 584–597] in The Extremophile Handbook (Springer,Tokyo). Horikoshi, K., Grant, W.R., Antranikian, G. & Robb F.T. (eds.) (2010).
Luo, H. & Robb, F. T. A modulator domain controlling thermal stability in the Group II chaperonins of Archaea. Arch. Biochem. Biophys. 512, 111–118 (2011).
Iizuka, R. et al. Characterization of archaeal group II chaperonin-ADP-metal fluoride complexes: implications that group II chaperonins operate as a “two-stroke engine”. J. Biol.Chem. 280, 40375–40383. Epub 2005 Sep 23. PMID, 16183634 (2005).
Booth, C. R. et al. Mechanism of lid closure in the eukaryotic chaperonin TRiC/CCT. Nat. Struct. Mol. Biol. 15, 746–753, 10.1038/nsmb.1436 (2008).
Ditzel, L. et al. Crystal structure of the thermosome, the archaeal chaperonin and homolog of CCT. Cell 93, 125–138 (1998).
Kurouski, D., Luo, H., Sereda, V., Robb, F. T. & Lednev, I. K. Deconstruction of stable cross-Beta fibrillar structures into toxic and nontoxic products using a mutated archaeal chaperonin. ACS Chem. Biol. 8, 2095–2101 (2013).
Yam, A. Y. et al. Defining the TRiC/CCT interactome links chaperonin function to stabilization of newly made proteins with complex topologies. Nat. Struct. Mol. Biol. 15, 1255-62, 10.1038/nsmb.1515 (2008).
Amit, M. et al. Equivalent mutations in the eight subunits of the chaperonin CCT produce dramatically different cellular and gene expression phenotypes. J. Mol. Biol. 401, 532–543, 10.1016/j.jmb.2010.06.037 (2010).
Kitamura, A. et al. Cytosolic chaperonin prevents polyglutamine toxicity with altering the aggregation state. Nat. Cell Biol. 10, 1163–1170, Epub 2006 Sep 17. PMID, 16980958 (2006).
Shahmoradian, S. H. et al. TRiC's tricks inhibit huntingtin aggregation. Elife 2,e00710, 10.7554/eLife.00710 (2013).
Hartman, D. J., Surin, B. P., Dixon, N. E., Hoogenraad, N. J. & Høj, P. B. Substoichiometric amounts of the molecular chaperones GroEL and GroES prevent thermal denaturation and aggregation of mammalian mitochondrial malate dehydrogenase in vitro. Proc. Natl. Acad. Sci. USA. 90, 2276–2280. (1993). Erratum in: Proc Natl Acad Sci USA 1993 Apr 5; 90, 3775. PMID, 8096339 (1993).
Kurouski, D., Luo, H., Sereda, V., Robb, F. T. & Lednev, I. K. Rapid degradation kinetics of amyloid fibrils under mild conditions by an archaeal chaperonin. Biochem. Biophys. Res. Comm. 422, 97–102 (2012).
Sun, Y., Makarava, N., Lee, C. I., Laksanalamai, P., Robb, F. T. & Baskakov, I. V. Conformational stability of PrP amyloid fibrils controls their smallest possible fragment size. J. Mol. Biol. 376, 1155–1167 (2008).
Luo, H., Zhang, P. & Robb, F. T. Oligomerization of an archaeal group II chaperonin is mediated by N-terminal salt bridges. Biochem. Biophys. Res. Commun. 2011 Sep 23; 413, 389–394, PMID, 21893040 (2011).
Boeckmann, B. et al. The Swiss-Prot protein knowledgebase and its supplement TrEMBL. Nucl. Acids Res. 31, 365–370 (2003).
Shomura, Y., Yoshida, T., Izuka, R., Maruyama, T., Yohda, M. & Miki, K. Crystal structures of the group II chaperonin from Thermococcus strain KS-1: steric hindrance by the substituted amino acid and inter-subunit rearrangement between two crystal forms. J. Mol. Biol. 335, 1265–1278 (2004).
Larkin, M. A. et al. ClustalW and ClustalX version 2. Bioinformatics 23, 2947–2948 (2007).
Figueiredo, L. et al. Functional characterization of an archaeal GroEL/GroES chaperonin system: significance of substrate encapsulation. J. Biol. Chem. 279, 1090–1099 (2004).
Bowers, K. J. et al. Scalable algorithms for molecular dynamics simulations on commodity clusters. Proceedings of the ACM/IEEE Conference on Supercomputing, Tampa, Florida, November 11–17, (2006). Proteome Res. 9, 1763–1771, 10.1021/pr900900x (2006).
Acknowledgements
This work was done under the umbrella of the agreement between the Euro-Mediterranean Institute of Science and Technology (IEMEST; Italy) and the Institute of Marine and Environmental Technology (IMET; USA) signed in March 2012. It was also partially supported by Grants AFOSR 03-S-28900 and 9550-10-1-0272 (FTR); and by the Air Force Office of Scientific Research under Grants AFOSR 03-S-28900 and 9550-10-1-0272 and the National Science Foundation under Grant No. CHE-1152752 (IKL); and a FFR2012, University of Palermo grant, CUP:B78C12000380001(AL). AJLM and FC were partially supported by IEMEST. This paper is IMET contribution 14-132.
Author information
Authors and Affiliations
Contributions
F.T.R., E. C.de M., A.J.L.M. and F. C. conceived and designed the experiments. A.L. and A.A.M. carried out molecular modeling and the simulation experiments. H.L., F.A. and F.T.R. designed mutant gene construction. W.K.M. and F.A. conducted recombinant protein expression and purification. W.K.M., F.A. and M.S. carried out protein characterization and enzyme stability-protection studies. M.S. and I.L. carried out fibril deconstruction experiments. F.T.R., E. C.de M., A.J.L.M. and F.C. wrote the paper.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Dataset 1
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Min, W., Angileri, F., Luo, H. et al. A human CCT5 gene mutation causing distal neuropathy impairs hexadecamer assembly in an archaeal model. Sci Rep 4, 6688 (2014). https://doi.org/10.1038/srep06688
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep06688
This article is cited by
-
Bridging human chaperonopathies and microbial chaperonins
Communications Biology (2019)
-
Structure of the human TRiC/CCT Subunit 5 associated with hereditary sensory neuropathy
Scientific Reports (2017)
-
Evolution of the archaeal and mammalian information processing systems: towards an archaeal model for human disease
Cellular and Molecular Life Sciences (2017)
-
CCT complex restricts neuropathogenic protein aggregation via autophagy
Nature Communications (2016)
-
Internal (His)6-tagging delivers a fully functional hetero-oligomeric class II chaperonin in high yield
Scientific Reports (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
